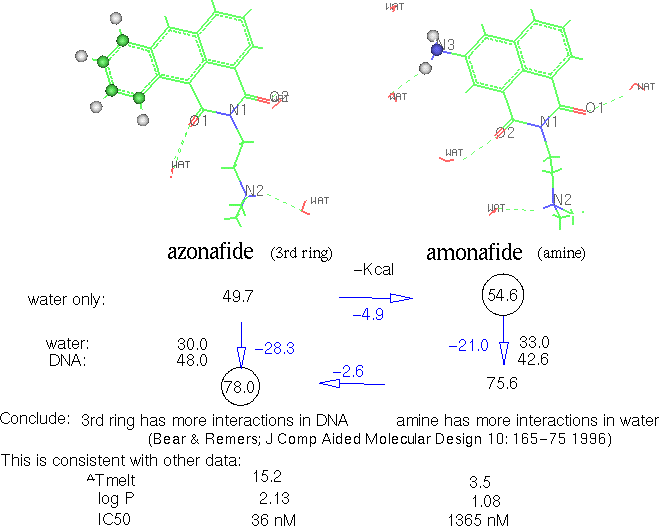
 Figure 22. Solvation
& intercalation relative energies of two compounds
Figure 22. Solvation
& intercalation relative energies of two compoundsINHIBITOR RESULTS AND DISCUSSION
Azonafide binds more strongly than amonafide (-57.3 vs. -47.9 kcal/mol respectively). Net enthalpy change (binding plus distortion in DNA and drug) favors azonafide over amonafide by -1.4 kcal/mol. Moving from solvent into intercalation has a net enthalpy loss of 25.5 for azonafide and 28.8 for amonafide. Combining the net drug-DNA binding enthalpy and the partial loss of solvation enthalpy gives a net enthalpy change for the overall process of -7.8 for azonafide and -3.1 for amonafide. Thus, azonafide intercalation is favored by -4.7 kcal/mol. This represents enthalpy and not free energy because we do not have quantitative estimates of the entropy changes resulting from intercalation. The larger azonafide would require more ordered waters and likely to have a greater entropy effect when they are released upon intercalation.
Figure 22. Solvation
& intercalation relative energies of two compounds
Atom by atom analysis of the interactions between intercalator and DNA shows that the difference of interaction can be accounted for fully by the third ring of azonafide vs. the amine of amonafide. The third ring consistently had a calculated interaction of 6-8 kcal/mol whereas the amonafide amine was only approximately 1 kcal/mol.
Dielectric setting affected hydrogen bonds. Distances of hydrogen to heavy acceptor atoms were approximately 0.3 A longer with dielectric 3 than the 1.6 A when dielectric 1 was used. The N to O distances opened a lesser extent, by 0.1 or less, toward the ~2.8 A measured experimentally in proteins. This would be because the reduced electrostatic interaction decreases the attraction.
Realistic dynamics were demonstrated by the wide movements of the drug side chain, transient breakage and re-annealing of internucleotide hydrogen bonds, and the second (opposite) docking attaining the position of the first. A measure of success in our method of simplifying the search of conformational space was the confirmation of conformation after a second docking in the opposite direction of the first result. In other words, if the first docking (started in the center) ended to the right, the second was done to the left to test if it would sweep all the way over to the right. In the major groove amonafide N2 coordinated with N7 and O6 of the nearest Guanine. The lowering to 41 Kcal net binding energy in one case was due only to distortion of a broken base pair at the end of the oligomer; intercalation of the drug was just like the 1st in all other ways.
It was observed both in these results and during the development of conditions that the side chain of azonafide had a marked propensity to rotate as long is it wasn't held by a hydrogen bond to a phosphate. In contrast, amonafide only had one case of marked side chain rotation. I propose this is because the larger chromophore allows the azonafide to reach out farther to allow freer rotation of the side chain.
Despite the difference in size of the chromophores of amonafide and azonafide the energy of interaction of intercalation is nearly the same in all four directions, 35-38 Kcal/mol. There is substantially more interaction by the 3rd ring of azonafide, 6-8, than by the amonafide amine, approximately 1. Major differences appear in the chain interaction with DNA. Results with hydrogen bonding to phosphate had 18-19 Kcal/mol whereas most others were around 10-12, with the notable exception of ecj35dd where coordination with N7-O6 conferred about the same amount as the phosphate.
The coordination with Guanine N7-O6 corresponds to calculations a decade ago predicting cations would be attracted to the N7 of guanine [Chen 1987]. The point is not so much the importance of the coordination as much that it illustrates the ability of the side chain to sweep around and allow an induced fit between the topoisomerase and DNA. The azonafide appears to have more possibilities of poisoning the enzyme due to a greater ability of the side chain movement.
Conclusions about amonafide vs. azonafide
Larger planar aromatic interaction provides stronger hydrophobic interaction with the base pairs it is intercalated between so it can also reach out a little, without falling out, allowing the side chain greater freedom of rotation.
The phosphate backbone, which is the target of the topoisomerase tyrosine, can be reached by intercalator side chain tertiary amine, which might possibly compete and interfere with the proton transfer necessary for release of the enzyme.
Error in force field based calculations
AMBER 3 force-field includes non-covalent parameters for electrostatic, van der Waals dispersion and hydrogen bonds [Weiner 1986 & 1984]. Not explicitly included are polarization and charge transfer, which would require quantum mechanics which is too complex for the macromolecules this study looks at. Although absolute Gibbs free energy cannot be determined, the similarity of the two intercalators allows consideration of relative binding.
The comparison obtained for azonafide provided a result which fits a reasonable qualitative sense of theory. However, the errors in the force field basis must be acknowledged. One of the first FEP calculations predicted solvation free energy of methanol/ethane within a Kcal/mol, 6.75 vs. 6.93, a 2.7% error [Jorgensen 1985]. Kuntz states "that force fields and empirical energy functions can rarely achieve better than +/- 2 Kcal/mol accuracy except within a family of compounds that have little conformational flexibility and that all bind in a very similar manner" [Kuntz 1994]. In favorable cases, relative free energies of association within 1 Kcal/mol of experiment have been achieved [Merz 1989]. However, inaccuracies in force fields and representation of the system and especially, sampling conformational space, have restricted the number of systems that could be done accurately [van Gunsteren 1990; Beveridge 1989]
Force fields are empirically based on solid packed crystals and not on isolated molecules. Protein parameters are based mainly on globular water soluble proteins whose structures have been successfully elucidated. Although modeling may appear to simulate motion of a single molecule, it is well to remember that the reference structure is as observed in a crystal, surrounded by and interacting with many solvent and buffer neighbors. This means that force-field parameters are conditioned by the crystal environment and any attempt to derive them from first principles must consider that environment [Boeyens 1996]. Force fields are generally based on crystal structures, which include environmental effects such as ion pairing, hydrogen bonds and co-crystallized solvent molecules. So an influence of these factors is implicitly included in the force field and it is therefore not appropriate to refer to molecular mechanics calculations as distinctly gas phase or in-vacuo even if not explicitly included in the optimization [Comba & Hambley 1995]. Optimized structures of naked molecules usually mimic crystal data pretty well. For small organic molecules, bond lengths are reproduced to within 0.005 A and angles to 1 degree [Allinger 1976] and for small coordination compounds to 0.010 A and 2-5 degrees [Bernhardt 1992, Comba 1993]. Dielectric constants include implicitly whatever is not explicitly dealt with in an electrostatic model. [Gilson 1995].
The hydrogen bond parameter is explicit in some force-fields (including version 3 but not version 4 of AMBER) yet the length of a hydrogen bond varies significantly depending on the environment. This reflects the lack of consensus on how to parameterize hydrogen bonds to reflect the real world variance. For OHO, reports of O to O distances range from 2.36 to 3.69 A and experimental bond energies from 31.5 to less than 1 kcal/mol. Analysis of Cambridge Structure Database shows that conjugated resonance also plays a role with O-O distances; from 2.39 for resonant intra-molecular B-diketone enols to over 2.80 for non-resonant crystals [Gilli 1996]. Hydrogen bonds of the same type in the same crystal vary 20% in contrast to covalent bonds varying only 2%. Hydrogen atom position usually inferred from point of heavy atoms and not necessarily placed well. Competition of solvent water reduces hydrogen bond strength [Jeffrey 1991]. Quantum calculations of 120 solutes indicated that hydrogen bonding is poorly correlated with electrostatics [Friesner 1998]. This sheds doubt on AMBER force-field which folded hydrogen bonding into electrostatic energies with ver 4 of that software. The comprehensiveness of a force-field parameterized for the hydrogen bond heavy atoms involved plus resonance with nearby groups affects the results.
Typically, around 95% of a calculation is spent on non-bonded interactions, which are based on gross approximation of a repulsive steric sphere surrounding an attractive/repulsive nuclear electrostatic point charge. This is a crude semblance of orbitals which actually undergo much polarization and distortion. It is ironic that so much computational time is spent on this rough approximation which is the source of much of the error.
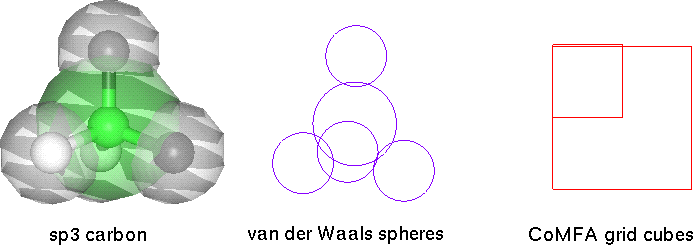 Figure 23.
Orbital approximations
Figure 23.
Orbital approximations
My study (while on summer internship at Abbott Inc.) comparing CoMFA (ligand only) with force field based minimization (receptor with docked ligand) for five enzymes (carbonic anhydrase, thymidine kinase, neuraminidase, thrombin, HIV protease) verified the weakness of time consuming molecular calculations. Essentially, the sphere and cube are equally poor approximations, while CoMFA is much faster to calculate.
Table 11. Error of CoMFA ligand vs. force field receptor
Carbonic Anhydrase |
Thymidine Kinase |
Neuraminidase * |
Thrombin |
HIV-Protease * |
|
| forcefield
target standard error |
0.39 |
0.63 |
0.90 |
1.13 |
0.74 |
| CoMFA ligand standard error |
0.33 |
0.56 |
0.59 |
0.94 |
1.10 |
| #inhibitors | 20 |
18 |
23 |
35 |
33 |
The CoMFA error was significantly (F test) greater for HIV Protease, less for Neuraminidase and about the same for the other three sets of enzyme inhibitors. The error was smallest for the set of small carbonic anhydrase inhibitors and largest for the set of large floppy HIV Protease inhibitors. The log order magnitude (10 fold) of the error makes it apparent that both methods are fraught with error and there is great need for improvements in binding prediction methodology.
Ludi design of additional azonafide analogs
Ludi was used upon several points of azonafide intercalated in DNA. A number of possibilities were generated that were provided to my advisor for exploration of synthetic feasibility.
SAR of topoisomerase inhibitors
Quantitative Structure Activity Relationship is a ligand centered approach involving statistical analysis of binding relative to chemical and physical properties of a set of compounds. It is both necessary and difficult, cheap and expensive.
The necessity is that bringing a drug into usage is enormously expensive. One approximation of the cost is that the combined government and industrial annual research is around $30 billion and there tends to be around 30 significant new drugs introduced per year. When you look at the time cost curve of a typical drug, it is apparent that clinical testing is where the heavy costs are and where there are many failures. It is important to spend enough on the early design stages, including modeling and QSAR, to help reduce the number of expensive clinical failures.
The difficulty is that inhibition depends on several subtle parameters, including steric fit, interactions of hydrogen bonding, charges, electron donating and accepting groups, hydrophobicity, and polarity. Since it is a statistical linear regression of a set of compounds, it can detect a trend but is poor at alerting one to interesting exceptions, outliers from the trend line. Those have to be ascertained by human reason. As noted above, topoisomerase is especially complex since there are essentially two receptors. How subtle the difference in activity is can sometimes be quite striking. There are some good examples among topoisomerase inhibitors.
The statistical analysis is relatively cheap and quick. However the necessary binding data requires synthesis of the compounds and measurement of activity, which can be costly.
SAR of azonafide analogs
Over 100 analogs of azonafide have been synthesized by the Remers group and assayed on cell cultures [Remers 1997].
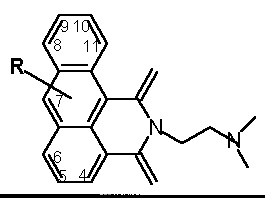 Table 12 Azonafide analog substituents
Table 12 Azonafide analog substituents
R |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
| -H | 1 | |||||||
| -C | 101 | 46 |
19 | 102 | ||||
| -CC | 15 | |||||||
| -N | 33/127 | 87 | 37 | 35 | 14 | 29 | 30 | 3 |
| -N(C)2 | 132 | |||||||
| -NCOC | 32 | 105 | 82 | 34 | 61 | 26 | 27 | 52 |
| -NCCOH | 89 | |||||||
| -NCOC(C)3 | 86 | 85 | ||||||
| -NCCN(C)2 | 72 | 47 | 62 | |||||
| -OH | 59 | 145 | 146 | 100 | 122 | 74 | 57 | |
| -OC | 71 | 54/81 | 20 | 103 | 133 | 70 | ||
| -OCC | 53 | 130 | 140 | |||||
| -OCCC | 135 | |||||||
| -OCCSC | 136 | |||||||
| -OCCN(C)2 | 104/138 | |||||||
| -OCCCOH | 137 | |||||||
| -SC | 141 | 25 | ||||||
| -SCC | 144 | |||||||
| -SO2C | 142 | |||||||
| -NO2 | 13 | 2 | ||||||
| -N=N | 88 | |||||||
| -Cl | 125 | 36/75/80 | 17 | 75 | 124 | 60/16/73 | 58 | |
| -I | 106 | 107 | 83 | |||||
| -F | 117 |
italics
-cyctosol localized; bold-nucleus localized [Mayr 1997]
To my novice eye this matrix of substituents appeared initially to be a good basis for a study of structure activity relations with the Apex and CoMFA software programs. Unfortunately, that was unproductive, presumably due to enormous variability of the whole cell assay. Literature searching about topoisomerase inhibitors revealed that many others have grappled with the difficulty of this topic. Hansch & Leo, the greats of SAR, said after analyzing a set of over 500 similar acting AMSA analogs that they are "so similar that they offer no interesting ideas for making more selective drugs" [Hansch 1995]. Yet, it is not quite so totally bleak. Some broad examination was done by spreadsheet analysis and literature searching, which turned up some trends and some ambiguities.
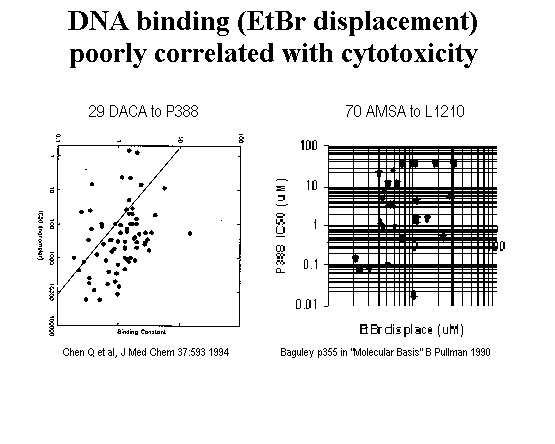
A graph of activity using averages of IC50 values for ovarian + melanoma cells and cardiotoxicity (LD50 to myocytes) indicates greater therapeutic indices with the most active compounds. This result suggests that the less active ones operate by a less specific and more generalized toxicity that affects all cells.
 Figure
24. Activity vs. cardio-toxicity graph (nM)
Figure
24. Activity vs. cardio-toxicity graph (nM)
Although the activities of this set range from 5-15,000 nM, most are in the range of 20-2000 nM. This three orders of magnitude is generally considered to be a minimum for any SAR study. Considering that the assay is based on whole cells rather than isolated enzyme, this set of compounds probably doesn’t span a wide enough range of activity for statistically significant analysis. Another indication that the activity range is too narrow is that by using the nuclear localization information (discussed next), a subset of 29 amino and alkyl analogs was run on Apex with the finding that a simple constant of -2.3 was sufficient to predict the activities with a poor but not horrible RMS of .82 (because the activity range for this subset is so narrow).
How flat is flat enough for intercalation?
There are some indications among the azonafides favoring flatness of the planar chromophore which would correspond to facilitating intercalation between DNA base pairs. The 6-amino analog is 61 fold more active than the bulkier 6-methyl amide. The shorter 6-dimethyl amine is 6 fold more active than the larger 6-dimethyl amine ethyl amine. The smaller 6-methyl is three fold more active than the 6-ethyl. The smaller 6-Cl is nearly four fold more active than the bulkier 6-I. It has been previously reported for a set of quinolones that flatness of the rings is correlated with substantial increase in topoisomerase 2 poisoning [Bryskier 1995]. The bulk of the alkoxy azonafides is not correlated with activity so some other factor may be at play for them.
Nuclear localization of amine and alkoxy analogs
Fluorescent microscopy with a subset of these azonafide analogs indicates that localization to the nucleus is favored by amine substituents and retarded by alkoxy groups [Mayr 1997].
The amines are generally quite active. It is possible that the amine helps mimic the properties of amino acids in order to be carried into cells via amino acid channels. The main exception is the 8 amino and that one is such an outlier from the others that suspicion is raised about experimental error or a unique mechanism.
The alkoxy analogs have a range of cytotoxicity similar to the amines but the poor nuclear localization and non-specificity of the whole cell assay leaves open the question about what mechanism is involved. Studies with other topoisomerase inhibitors have indicated some negative impact by alkoxy substituents. Methoxy azatoxins lack activity in a direct topoisomerase-DNA assay [Tepe 1996]. HIV integrase inhibitors, which are likely to need nuclear localization, are made less active by methoxy [Fesen 1994]. It has been reported for a set of aza acridines that 7 oxy is only 1/3 as active as the 7 hydro [Chen 1994]. It is also reasonable to think that an electron rich oxygen on the aromatic rings facing the direction of intercalation might be repulsive with respect to electron rich DNA and therefore have a smaller fraction intercalated. There is more to understand about the strength of activity of the alkoxy effect and of the act of intercalation.
The nitro advantage?
The two azonafide analogs with nitro substituents were highly active, consistent with finding reported with other nitro-aromatics. Mitonafide (3-nitro) was around 10 fold more potent than amonafide (3-amino) in vitro cell cultures [Brana 1995 & 1981] and causes a greater amount of topoisomerase II mediated DNA fragments [Hsiang 1989]. If only a portion of the 10 fold difference can be explained by the DNA affinity, this implies some other mechanism is confounding the data: whether it be covalent reaction or greater cell take-up or other mechanisms. NO2 substituent is more active at P388 cell killing than nitrogen in the ring at the same position of set of acridines [Chen 1994].
The nitro electron withdrawing property was hypothesized to increase intercalation interaction with nucleotides. While a DNA unwinding gel shift indicates little difference [Brana 1995], more precise Tmelt measurements show a greater delta Tm of 6.6 for the nitro vs. 3.5 for the amino version [Solyom] indicating modestly more intercalation. Analysis of 188 nitro-aromatics found that ones with three or more fused rings are 10-100 times more mutagenic than expected by log P and ELUMO (electron withdrawing groups) [Sams 1995] supporting a hypothesis of metabolic reduction leading to DNA adducts [Einisto 1991; Bryant 1991].
CN and NO2 confer considerable cytotoxicity on several classes of compounds but action is likely to be non specific alkylation of more than just topoisomerase since it is not clear that the planar aromatic portion is a very effective means of targeting to topoisomerase.
Odd over even
There is a trend for odd numbered positions to be better than even numbered ones, no matter which substituent. There is no clear reason for this except that the 5 position appears to be favorable and may bias the statistics.
A broader look at other topoisomerase inhibitors
The mutation structure analysis shown earlier helped introduce the subtle differences between compounds which have previously been clumped together as one big group of planar aromatic topoisomerase inhibitors. The correct categorization and distinction of these agents involves a great deal of subtlety and ambiguity. Even for similar compounds having the characteristic planar composition for intercalation
There is little reason why other enzymes which act on DNA wouldn’t be affected by the intercalators so there is great question about just what is being measured in whole cell assays. Such other possibilities would include helicase, which has similar function to topoisomerase and might even be required for topoisomerase trapping [Howard 1994]; the DNA methyltransferases, for which association with tumorigenicity and apoptosis have been shown [Baylin 1998]; Poly(ADP-ribose) polymerase (PARP), which is in the apoptosis signaling pathway and is inhibited by amino-naphthalimide which has structure similar to the amonafide discussed in this paper [Banasik 1992].
Synergy and antagonism of topoisomerase inhibitors
A mixture of topoisomerase 1 inhibitor with different types of topoisomerase 2 inhibitors found synergy, especially when topotecan is followed by etoposide [Grabowski 1996]. CHO cells allowed to repair after UV induced pyrimidine dimerization, were not affected by inhibitors of either topoisomerase 1 or topoisomerase 2 but when both inhibitors were used simultaneously the DNA repair is markedly inhibited. In cells deficient for topoisomerase 1, repair is inhibited by topoisomerase 2 inhibitor alone [Stevnsner 1993]. This suggests therapeutic a advantage in blending inhibitors of both topoisomerase 1 and 2.
It is tempting to think that using a mixture of anti-topoisomerase 2 agents having different mechanisms would provide synergy but there are some cases where the opposite is clearly the case. Merbarone inhibits topoisomerase 2 unwinding (IC50 20 uM) without topoisomerase associated DNA fragments yet it strongly interferes with m-AMSA or teniposide induced damage, indicating a different mechanism [Drake 1989b]. Bis-dioxopiperazines inhibit topoisomerase 2 unwinding (IC50 13 uM) without fragmentation yet strongly interfere with etoposide induced DNA fragmentation [Tanabe 1991]. Aclarubicin (a tri-glycosylated anthracycline) is weakly cytotoxic yet at 10 nM it strongly interferes with m-AMSA and modestly with etoposide cytotoxicity [Jensen 1990]. Of 5 cytotoxins which don’t themselves cause topoisomerase associated DNA fragments, 4 do strongly interfere with teniposide induced damage [Chen 1993].
Problems with topoisomerase inhibition strategy
Like many other anti-cancer agents, the topoisomerase inhibitors are a double-edged sword and may themselves cause unwanted mutation and cancer [Anderson 1994]. They are also able to induce drug resistance [Giaccone 1992] which would cause refractory remission requiring even more toxic therapies. Leukemias with abnormalities in chromosome 11p23 are not uncommon after treatment with topoisomerase II drugs [Cortes 1994]. The NCI screening program found some sensitivity to topoisomerase inhibitors in ras mutated cells, yet ara-C was even more effective for those cells [Koo 1996] The importance of an intact p53 apoptotic pathway was discussed in the introduction. One medical professional has pointed out that decades of research on new treatments has had little impact on reducing cancer deaths and a shift to prevention and early detection is likely to be more cost effective [Bailar 1997]. Nonetheless, there are situations and cell types which lend themselves to topoisomerase poisoning therapy.
Conclusions, implications and suggestion for future study
It is sensible to counter the growth of cancer with agents specific to particular proteins which are expressed excessively. As indicated in introduction there are some cancers high in topoisomerase 2 and those situations would be the greatest candidates for the inhibitors discussed in this paper. Co-amplified receptors offer the possibility of selective targeting to transformed cells. Although there is much more to learn about the interaction of the three topoisomerase genes with their neighboring oncogenes it is evident that all three are from chromosomal locations with known susceptibility to damage leading to cancer.
Study of topoisomerase poisoning is particularly difficult because two receptors, enzyme plus DNA, are involved. Study of the structure of topoisomerase 2 indicates that the widely accepted model of 5’ overhang is inconsistent with topoisomerase structure and that 3’ overhang is more likely. A major groove aligning helix has been identified near the active site. Homology modeling shows that the active site region is well conserved between yeast and humans with some exceptions:
Drug resistant mutant structure analysis suggests multiple sites of action by topoisomerase inhibitors and suggests the region of DNA binding
Prediction of intercalator binding of amonafide and azonafide to DNA is correctly ranked by molecular mechanics. However, there remains significant error from experimental measures. Force field error derives from parameterization from a limited set of packed soluble crystals; the approximation of electron orbitals as a hard van der Waals sphere unable to polarize; and poor handling of hydrogen bond variability. The larger set of azonafide analogs had too much variability on whole cell assays and insufficient range of activity for statistically significant QSAR, but some general observations are possible. An effective intercalating topoisomerase 2 poison has 3-4 planar rings with at least one amino group which can move about in the major groove and interfere with the normal function of residues of topoisomerase near the active site tyrosine.
Measurements of topoisomerase 2 inhibition need to be done on a simple enzyme-DNA assay, preferably with separate testing for purified types 2
a and 2b , for improved QSAR data.
Copyright © 1998 Soaring Bear;
Please send your comments bear@dakotacom.net