INTRODUCTION
Each year there are 1 ¼ million diagnoses and a half million deaths per year from various types of cancer. That’s 1500 per day or 1 every minute [ACS 1995]. Every one of us has been affected by the sickness and costs of our relatives, friends and neighbors.
Table 1 Cancer incidence
annually |
Diagnoses |
deaths |
survival % |
| lung | 170,000 |
157,000 |
7 |
| colon | 100,000 |
48,000 |
52 |
| breast | 183,000 |
46,000 |
75 |
| prostate | 244,000 |
40,000 |
83 |
Smoking accounts for 87% of lung cancers and about 1/3 of all cancer deaths. While lifestyle can prevent some cancers, the death rate is a continuing reminder of the tremendous task of finding more effective treatments. In addition to cancer, the cytotoxins discussed below also have application as antibiotics and against parasites.
Topoisomerase targeting of chemotherapy
One of the targets for chemotherapy is topoisomerase, a class of enzymes in the nucleus of all living cells, which affect the topological structure of DNA. Their action of temporarily cutting the DNA to allow winding and unwinding to adjust the amount of kinking (or topology) is critically important to transcription, replication and chromosome structure. Cells die when topoisomerase is inhibited. This paper presents and discusses advances in understanding how topoisomerase poisons work.
EC classification ambiguity
Although topoisomerases have been categorized in the widely used Enzyme Classification (EC) system as 5.99.1.x, the complexity of function of topoisomerase actually could allow it’s inclusion in any of the other 5 major classes as well.
The EC number 5.99 classifies it as 'other isomerases' (group 5.99) along with only one other enzyme, thiocyanate isomerase, but it’s function is not really a change of isomer; there is simply a change of twisting. It is similar to hydrolases (group 3) during the first half of its cycle. Using a temporary tyrosine hydroxyl in place of a permanent water to break the phosphate bond is the only subtlety that distinguishes topoisomerase from similar acting restriction endonucleases and hydrolases (group 3). Another indication for group 3 is that a simple L43K mutation in the DNase, NaeI, (EC 3.1.21.4) gives topoisomerase activity [Jo 1996]. The lysis of the phosphodiester bond during the first half of its cycle makes it a lyase (group 4), similar to apurinic endonuclease (4.2.99.18). It is certainly a ligase (group 6) during the second half of its cycle when it rejoins the broken DNA. The proton transfer (from tyrosine to the DNA free end hydroxyl) during trans-esterification could allow it to be called an oxido-reductase (1). Transferring a tyrosine temporarily to the DNA phosphate might qualify it as a transferase (group 2).
Table 2 EC classification of topoisomerase
| # | EC class |
definition |
why topoisomerase fits |
| 1 | oxidoreductases | oxidation reduction and electron or proton transfer | proton transfer |
| 2 | transferases | groups are transferred | tyrosine transferred to DNA |
| 3 | hydrolases | hydrolytic cleavage of covalent bonds | tyrosine hydroxyl breaks phosphate like endonucleases in this group |
| 4 | lyases | form/break double bonds | lysis of phosphodiester bond |
| 5 | isomerase | isomerization | only twisting change |
| 6 | ligase | covalent joining | re-ligation of broken DNA |
The complexity of topoisomerase, which makes for difficulty in categorizing it, is a theme that will be seen throughout this paper in trying to understand it for designing better and specific inhibitors.
Three types of topoisomerase in humans
Simpler organisms have topoisomerase 1 (EC# 5.99.1.2) which makes one cut. Higher organisms have, in addition, topoisomerase 2 (EC# 5.99.1.3) which makes two cuts, 4 base pairs apart (similar to many restriction enzymes.) Types 1 and 2 have some degree of redundancy in function [Garinther 1997] and are induced when one is inhibited [Whitacre 1997] but each is critical enough that any disruption of either of them shifts the balance towards more apoptosis. Synergistic cytotoxicity is obtained when both are inhibited [Grabowski 1996]. Both types 1 and 2 cut DNA by tyrosine attack of the phosphodiester bond of DNA (discussed below).
In humans, three known types are 1, 2
a and 2b ; made by three separate chromosomes, 20q11.2, 17q21 and 3p24, respectively. 2b is highly homologous to 2a (but with some key differences discussed later) and expression is induced by damage to 2a [Khelifa 1994; Boege 1993], so 2b appears to be something of a backup for cells. Anthracycline treatment was associated with increased 2B in 9 relapsed leukemia patients [Gieseler 1997]. There has been some evidence that functional loss of one can be partially substituted by another [Stevnser 1993]. 2a and 2B are similar enough to form a substantial fraction of alpha/beta heterodimers [Biersack 1996]. Doxorubicin resistant cells have 4 fold expression of topoisomerase 1 [Riou 193]. More comparison of 2a and 2B will be presented in the sequence analysis section.
Table 3 Differences between topoisomerase 1, 2
a & 2b1 |
2 a |
2 b |
|
| function | remove negative supercoils | ||
| cell cycle | fairly constant but higher in S phase during transcription LiuWang 87 | gradually increasing during S phase to a peak at gap 2, just before mitosis replication Adachi low in differentiated cells Bauman | constant Woessner92 |
| location along DNA | transcribing areas | uniform | |
| during interphase | nucleus Meyer 1997 | spotted Meyer 1997
|
reticular |
| during mitosis | distributed through chromosomes Meyer 1997 | chromosome axes centriols Meyer 1997
|
cytosol |
| gene | 20q11.2-13.1 Kunze 89 | 17q21-22
|
3p24 Jenkins92 |
| nearby oncogene | src Hollings 94 | erbB2 & RAR
|
MLH1, erbB4, hOGG1 |
| protein size (human) | 765 (91 kd) monomer | 1530 (174 kd) dimer |
1626 (180 kd) dimer |
| # cuts | 1 | 2 (4 bp overlap) | same as 2a |
| ion | Mg (2mM) Zhu | Mg (7.5mM) osheroff87 | |
| pH opt | 8.9Gieseler 97b | 7.9Gieseler 97b | |
| energy | none | ATP to re-ligate | " |
| inhibitors | camptothecin WallWani66 quinolones Wrigley flavones Boege96 |
2-4 planar rings | |
| inhibition | cpt: late S accumulationInaba | etop: G2 arrestInaba |
DNA is organized in loops which are fixed to the nuclear matrix at matrix attached regions [Tsutsui 1988]. Topoisomerase 2a is a major nuclear scaffold protein and is concentrated at the attachments of these loops [Gasser 1987].
Oncogenes near topoisomerase genes help targeting
Investigation of the gene locations for the DNA which codes for topoisomerase reveals that all three human topoisomerase genes are near cancer associated oncogenes. Literature search reveals evidence that topoisomerase is co-amplified in some oncogene associated tumors. Antibodies directed at the products of the oncogenes might help target topoisomerase inhibitors to cancerous cells.
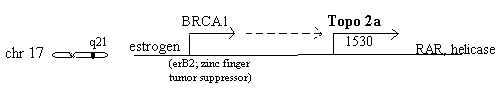
Chromosome 17q21 - topo 2a, erbB2 & RAR
The 2a gene is very close to erbB2 (also known as BRCA1 or HER2) the widely discussed breast cancer gene on chromosome 17q21 which is stimulated by estrogen. Estrogen stimulation of human breast cancer cells also induces topoisomerase 2 synthesis [Epstein 1989]. A study of 117 breast cancers found 25 (21%) having over 2.3 times normal erbB2 and of these, three (12%) were also high in topoisomerase 2, in contrast to zero of the other 92 [Smith 1993]. A different study of 230 breast cancers found topoisomerase 2a elevated in 11%, but undetectable in nonmalignant breast epithelium [Jarvinen 1996]. A lung adenocarcinoma line, Calu3, possesses co-amplified erbB2 and topoisomerase 2a [Keith 1992] indicating this relationship may be broader than just breast cancer. It has been "predicted that tumors which exhibit co-amplification of erbB2 and topoisomerase 2a may benefit from treatment with topoisomerase 2 inhibitory drugs" [Murphy 1995].
ErbB2 p185 protein has been observed in breast, stomach and ovarian cancers and appears to be inversely correlated with survival [Slamon 1989]. The fact that both alleles of erbB2 must be inactivated for cancer and the presence of a putative zinc finger (typical of DNA binding) of this nuclear protein suggest it is a growth/tumor suppresser [Futreal 1994, Giunciuglio 1995, Neuhausen 1994]. High erbB2 is associated with reduced benefit from megestrol or fadrozole hormone therapy and with poorer prognosis [Leitzel 1995] indicating the need for other therapeutic strategies. It is an EGF class trans-membrane receptor and tyrosine kinase. It is very low in normal tissue [Press 1990] and 10-100 fold higher on the surface of tumor cells [Kraus 1987, Berger 1988, Paik 1990]. Its extra-cellular accessibility and growth promoting activity make it a very attractive target for antibody blockade [Wels 1996]. Antibodies to erbB2 surface receptor have had some success in animal models, and most notably, the combination of 1 ug antibody with 43 ug/Kg of doxorubicin (the topoisomerase 2 inhibitor) had greater effect on human stomach cancer cells than either of the treatments alone [CR King 1996]. In view of the long hope of antibody targeting of cytotoxins this combination could be taken a step further in forming a conjugate with a hydrolyzable linker that would release the cytotoxin to the tumor region.
Retinoic acid receptor alpha is another gene nearby on chromosome 17 and can be co-amplified with erbB2 and topoisomerase 2a [Keith 1993].
Chromosome 3p24 - topo 2B, MLH1, hOGG1, erb4B, LOH
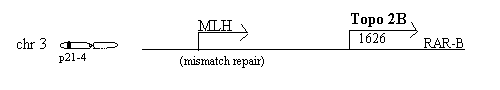
The topoisomerase 2B gene at chromosome 3p24 [Jenkins 1992] is near the gene for MLH1, a mismatch repair gene on 3p21.3-23 [Bronner 1994] Mutations in MLH1 are associated with 1/3 of hereditary non-polyposis colorectal cancer [Hemminki 1994]. The MLH1 gene encodes a protein of 756 residues, 84.6 kD, localized in the nucleus, which binds to mismatched DNA sequences and is associated with G2 arrest [Prolla 1996]. It is interesting to speculate how valuable a binder of mismatched DNA might work with topoisomerase during the time that the DNA is cut. Cisplatin can select for reduced hMLH1 and a MLH1 deficient cell line, HCT116, is 2 fold resistant to cisplatin [Aebi 1996]. This suggests a limit to usefulness of cisplatin and the value of multiple mechanisms of chemotherapy, including topoisomerase inhibitors.
hOGG1, human oxo-guanine glycosylase, another repair enzyme, is encoded at 3p25/26, a region commonly deleted in cancers [Lu 1997]
Erb4B is also nearby, at 3p22-24.1 [Drabkin 1988]
Distinct homozygous deletion at the nearby 3p21.3 is a critical event in the pathogenesis of lung cancer [Roche 1996]. Of 49 non small cell lung carcinomas examined in one study loss of heterozygosity, LOH was found in 3p21 in 13 of 30 and more specifically at the MLH1 locus in 9 of 11 (82%) of informative cases [Wieland 96]. Loss of heterozygosity on the 3p gene at the D3S2 locus has also been observed in a large percentage of small groups of cervical cancer patients [Yokota 1989, Karlsen 1994, Magnusson 1996] and deletions of that region appears to be a higher risk factor for cervical cancer than HPV [Yokota 1989].
Chromosome 20q11 - topo I, src
Topoisomerase 1 gene on chromosome 20q11.2-13.1 is near one of the src signal transduction tyrosine kinase family, an intensively studied group of oncogenes [Kunze 1989, Hollings 1994, Parsons 1997] two of which have recently been crystallized [Sicheri 1997]. Substantial deletions in this region are associated with tumors [Asimakopoulos 1994].
Targeting high levels of topoisomerase 2
Because topoisomerase 2 drugs act as poisons rather than as classical competitive inhibitors the action is often directly proportional to enzyme levels in cells. Rapidly proliferating cells, which contain relatively high topoisomerase 2 [Nelson 1987, Hsiang 1988] are very sensitive to the poisons [Sullivan 1987]. Differentiated cells have relatively low topoisomerase 2 levels and are resistant to the poisons [Kaufmann 1991]. Tumor cells selected for poison resistance often have low topoisomerase 2 levels [Webb 1991] although this is not always the case [Campain 1995]. Conversely, cells expressing topoisomerase 2 (by selection or transfection) are very sensitive to the poisons [Davies 1988, Nitiss 1992].
Some ovarian tumors can be extremely high (10 fold) in topoisomerase 2 or topoisomerase 1 [van der Zee 1991] and topoisomerase 2a is higher in malignant than benign tumors [Cornarotti 1996]. Topoisomerase 2 was found high in Dunning rat prostate adeno-carcinomas and level correlated with tissue growth rate [Nelson 1987]. non-Hodgkin’s lymphoma tissue of 36 patients was higher in topoisomerase 2a mRNA in the more advanced cases [Lohri 1997]. Lung tumors from 20 patients were high in topoisomerase 2 [Koomagi 1996]. A topoisomerase 2 defect has been reported in Li Fraumeni syndrome, an inherited susceptibility to a variety of cancers [Cunningham 1991]. Sensitivity to topoisomerase inhibitors is greatest during the proliferative mid-log stage of cell culture [Sullivan 1986]. Topoisomerase 2 is high in HL-60 human progranulocytic leukemia cells and decreases substantially when induced to mature [Kaufmann 1991]. CHO cells selected for topoisomerase 2 sensitivity develop 3 fold expression and are hypersensitive to amsacrine [Davies 1988].
Cells transfected with high levels of topoisomerase 2a are resistant to platinum therapy, presumably because DNA topology adjustments facilitate access to repair enzymes [Cornarotti 1996]. When cells are high in topoisomerase, it makes simple logical sense to treat with topoisomerase targeting agents. Heregulin transfected cells become exquisitely sensitive to doxorubicin and etoposide, presumably by up-regulation of topoisomerase 2 via the erbB2 promoter (discussed below) [Lupu 1996]. A different line of evidence for the value of inhibiting topoisomerase was with anti-sense gene constructs targeting topoisomerase 2a which inhibited prostate cancer cells by 39% [Lee 1996].
Bone marrow of healthy volunteers contain about the same levels of topoisomerase 2 as the HL-60 leukemia [Kaufmann 1991] so there is the old dilemma of cancer drug toxicity to healthy cells as well as to tumor cells which limits use of topoisomerase poisons to special situations such as when topoisomerase is high. Reduced levels of topoisomerase 2 mRNA is one of the mechanisms of drug resistance [Wang H 1997, Withoff 1996, Houlbrook 1996] and could be considered a contraindication for the value of topoisomerase inhibiting drugs.
Heat is one of the few known stimulators of topoisomerase 2a levels. Human epidermoid cancer cells exposed to 42 C for 3 hours resulted in a five fold increase of the mRNA. Shorter heating periods also had an increase and enhanced sensitivity to etoposide as well [Matsuo 1993]. Hyperthermia has been experimented with as a cancer therapy and it appears that topoisomerase poisons would be a sensible adjunct. [Sherar 1997; Bisht 1996]
Immediately upstream of the human topoisomerase 2a gene, at -37 to -10 b.p. are two DNA segments identified as potential binding sites for cMyb, the hematopoietic transcription factor associated with cell proliferation plus a possible site for c-Myc [Fraser 1995, Hochhauser 1992]. Transfection of c-myb increases topoisomerase 2a [Brandt 1997].
Cases of cancer where topoisomerase 2 is high are particularly good candidates for prescribing topoisomerase 2 poisons.
How inhibition of topoisomerase 2 kills cells
Topoisomerase induced damage is a complex process involving fundamental cell cycle biochemical pathways. Understanding this is important to understanding some of the reasons cells are either more or less susceptible to topoisomerase inhibitors.
Evidence for this model is based on a piecemeal assembly of many molecular biology studies. It serves to show the complexity of events when topoisomerase is inhibited. The gradual increase of the level of topoisomerase 2 during S phase is indicated by a widening band in the cell cycle circle. [Sullivan 1986]. The three key components for conveying the topoisomerase inhibitor damage are p53, Bax and PARP. A number of other modulatory factors also have roles.
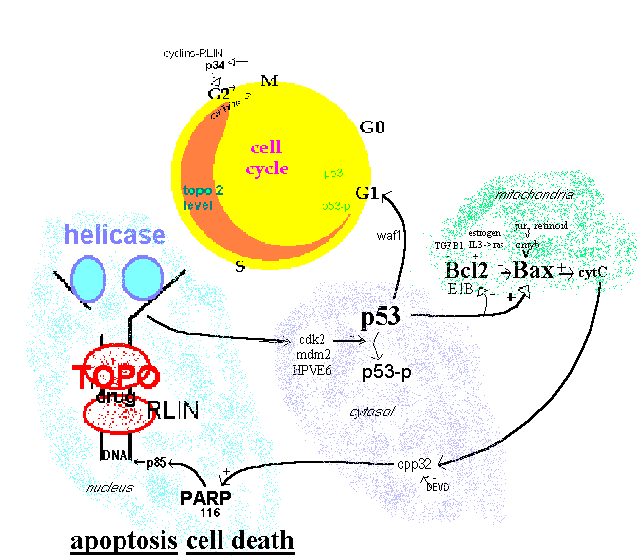 Figure 1.
Topoisomerase effect on cell cycle
Figure 1.
Topoisomerase effect on cell cycle
Intact P53 is required for G1 arrest by cytotoxins including the topoisomerase inhibiting etoposide and doxorubicin [Fan 1994; Lowe 1993]. P53 phosphorylation and cell viability is decreased by the topoisomerase inhibiting ellipticine [Ohashi 1995]. P53 can down-regulate expression of topoisomerase 2a promoter (attached to luciferase) by 15 fold [Wang Q 1997] indicating a feedback loop which may be difficult to disentangle. P53 mutant cells are resistant to doxorubicin but maintain sensitivity to cis-diamminedichloroplatinum [Vikhanskaya 1995]. It can be concluded from this evidence that tumors with mutant P53 are not good candidates for topoisomerase inhibiting agents.
The Bax:Bcl2 ratio needs to be high for etoposide induced apoptosis[Chresta 1996] and raising the ratio by antisense oligomers to Bcl2 mRNA is a strategy being investigated by Genta Inc. of San Diego [Rawls 1997]. Cells augmented with transfected Bcl2 cDNA had reduced etoposide induced DNA crosslinks and apoptosis [Lock 1996, Kamesaki 1993]. The ratio is regulated by p53 [Miyashita 1994; Naumovski 1996].
PARP activation to the p85 endonuclease is inhibited by Bcl2 [Bonfoco 1996, Messmer 1996]. PARP inhibition by 3-aminobenzamide blocks etoposide induced apoptosis [Bernardi 1995].
It can be seen in this section that intact apoptotic signaling pathway is necessary for effectiveness of topoisomerase 2 targeting agents.
In evolution there is a continuous interplay of offense and defense. The key role of adjusting DNA makes topoisomerase a natural target of chemicals to kill invading pathogens. Some structural differences between species could provide a basis for species selectivity. Inhibitors naturally produced by plants include berberine, ellipticine, podophyllotoxin, lapachone, and camptothecin. Many commercial dyes are planar aromatics which can have some topoisomerase inhibition and design of new ones take this into account by making the compound large or floppy enough to reduce the intercalation ability [DeVito 1996].
Complexity of dual receptors
Topoisomerase 2 inhibition is more challenging, and interesting, than the usual ligand-receptor research due to the complexity of interacting with two receptors at the same time. Most enzyme inhibitors that have been studied involve a compound docking into an active or nearby allosteric site to interfere with entry or reaction of the normal substrate. Inhibitors of the topoisomerase 2 cleavage complex can be looked at as having two parts: the aromatic part that intercalates between DNA base pairs and a more polar portion that interacts with topoisomerase. Simple intercalation is not enough to inhibit the complex.
Ordinary single topoisomerase dual receptor-ligand
 Figure 2. Dual
Receptor complexity
Figure 2. Dual
Receptor complexity
A small compound of just 30-50 atoms or 0.5 kD is like a puny flea or small fishhook that snags together and devastates two large elephants: the huge complex of topoisomerase 2, composed of around 20,000 atoms or 170 kD, plus around 30 BP of DNA composed of 2000 atoms or 12 kD.
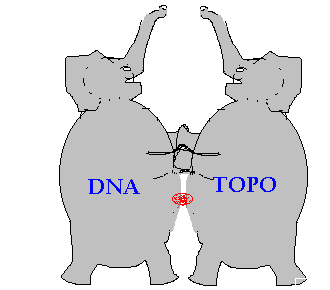 Figure 3.
Inhibitor is like a fishhook snagging two elephants
Figure 3.
Inhibitor is like a fishhook snagging two elephants
The intercalator doesn't keep topoisomerase from attaching to DNA but does prevent topoisomerase from reconnecting the DNA and detaching. So the inhibitor acts like a glue or a fishhook that holds the topoisomerase 2 attached to the DNA. Other cellular factors (i.e. P53 pathway) detect this damage and send the cell into apoptotic death.
topo--topo.DNA---rotate---DNA.topo--X | v apoptosis
Figure 4. topoisomerase-DNA fixation kills cell
Designing better topoisomerase trapping agents
Computational chemistry has the promise of detecting trends and predicting activity of unmade compounds to help prioritize which ones should be made first. There have been attempts to identify structure activity relationships but only mixed results. Structure activity relationship was investigated by me in stages:
Azonafide has a modest modification on the structure of amonafide. The former has a third ring replacing the amino group of the latter. A comparison of the relative DNA binding strengths of these compounds was calculated by molecular dynamics using force-fields. DNA binding is necessary but not sufficient for this class of topoisomerase inhibitor. Since the bulk of the compound is bound by DNA and the structure of DNA is well established, we sought to estimate the change in binding affinity caused by the modest change in the structure of amonafide.
This work focuses specifically on topoisomerase 2 inhibitors which allow the first step of cutting DNA but freeze the transition "cleavable complex" leading to apoptotic cell death. A structural composite of these compounds includes two common domains: 1) a flat planar polycyclic molecular surface which intercalates between base pairs of the DNA, and 2) a side chain that extends into the major or minor groove and can move around. Millions of doses of topoisomerase 2 inhibiting anthracyclines (e.g. doxorubicin) have been administered against cancer with a modicum of success. Such compounds work by trapping topoisomerase onto DNA hence driving cells into apoptotic death. However, anthracyclines also readily form radicals that cause heart and other damage [Dorshow 1980; Gianni 1982]. Even though this toxicity has been recognized for two decades and huge sums of money has been poured into research, exemplified by 2582 abstracts in Medline since 1966, this problem has not been overcome [Shan 1996]. The work presented in this paper is part of the effort to design new drugs having a similar action on topoisomerase without the radical damage caused by anthracyclines. Additional considerations are targeting specifically to cancer cells, avoiding healthy cells, and avoiding multi-drug resistance.
It was understood as early as 1961, with brass rod models, that the planar aromatic AMSA intercalates between bases of DNA [Lerman 1961]. That was only a few years after DNA structure was determined and well before topoisomerase was identified. And yet, intercalation is insufficient for trapping topoisomerase 2 as indicated by several lines of evidence. ortho-AMSA is a slightly better intercalator than meta-AMSA [Pommier 1987] but virtually inactive as a topoisomerase poison [Zwelling 1981, Nelson 1984]. A lack of correlation between intercalation and topoisomerase inhibition is also reported with anthracyclines [Capranico 1990] and amsacrines [Cain 1975; Wilson 1981; Wadkins 1989] although a weak correlation was reported by one group for amsacrines [Baguley 1990]. Displacement of ethidium bromide by 29 DACA analogs is un-correlated with P388 cell killing [Chen 1994]. DNA binding vs. activity is not well related and this helps to demonstrate that DNA binding is necessary but not sufficient. The compound presumably must additionally have appropriate interaction with topoisomerase to have good activity.
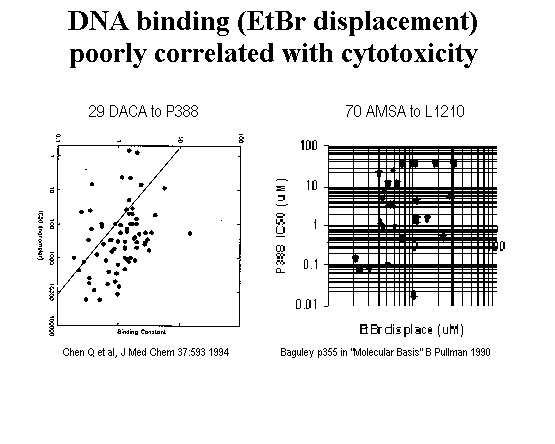 Figure 5.
DACA intercalation unrelated to cell killing (Chen 1994 data)
Figure 5.
DACA intercalation unrelated to cell killing (Chen 1994 data)
In contrast to the permanent hydrolytic cuts made by restriction endonucleases, topoisomerase cuts are temporary, and the enzyme must protect the loose ends from undesirable recombination until the reaction is reversed to re-ligate the break. The DNA super-coiling becomes either slightly tighter or looser, which is a difficult endpoint to measure. It is observed with gel mobility assays in isolated closed circle PBR322 DNA or kinetoplast but not with normal linear DNA. Hence, it is harder to follow the kinetics of topoisomerase than of simpler endonucleases. Because the cleavage complex trapping type of inhibitors which are discussed later allows the first trans-esterification, which cuts DNA, but prevents the second trans-esterification, which re-ligates, it is worth looking closer at this reaction.
Kinetics of topoisomerase winding of DNA
The kinetics of the temporary cutting topoisomerase has been contrasted with permanent cutting endonucleases in a simplified way:
endonuclease: E + S <> ES <>> ES* > E + S*
topoisomerase: E + S <> ES <> ES*
<<> E + S*where E= enzyme, S= DNA substrate, S*= cut DNA)
The difference is in reversibility with topoisomerase finishing like it started [Osheroff 1989], albeit slightly tighter or looser in super-coiling.
A more accurate representation for topoisomerase 2 has been described as:
KS Krelax
processive Krelax Krelax
E + S <> ES ----> ES-2 >>>>>> ER+2 -----> ER <-----> E + R
^ ^
v Koff v
E+S-2 E+R+2
where ES-2=supercoiled minus 2 turns or 1 loosening by topoisomerase
R+2=fully relaxed plus 2 turns [Osheroff 1983]
DNA can get relaxed by 1 turn (S-2) per step until fully relaxed (R) or past that to super-coiling in the other direction. Generally, the rate of relaxation Krelax must be faster than the rate of dissociation Koff. Since topoisomerase is present in catalytic amounts and must dissociate from relaxed DNA to go on to the next super-coiled site, the rate of relaxation must be related to the dissociation of the ER complex.
Conditions which make it less processive (decrease Krelax or increase Koff and reduce the amount of relaxed DNA), include high ionic strength (>175 mM), high Mg2+, (>15 mM), high pH >10. [Osheroff 1983]
The states of the topoisomerase tyrosine attachment and release is shown by ab initio calculations in this work to have nearly the same energies.
Categorizing topoisomerase inhibitors
A number of topoisomerase 2 inhibitors have been identified and developed including anthracyclines, anthrapyrazoles (mitoxantrones), acridines, benzophenazine, 4’-O-demethyl-podophyllotoxins, ellipticines, azatoxins, carbolines, batracylin, benzo-napthofuran, napthalimides, fagaronin, and intoplicines. With this growing mass of data, there have been some recent crude attempts to categorize topoisomerase 2 inhibitors into mechanistic classes. One recent review simply points out that topoisomerase 2 inhibitors "can be roughly classified into two groups: intercalators and non-intercalators [Wang H 1997]. One of the deans of topoisomerase research, Yves Pommier [1997] at first says there are two groups: those that trap cleavable complexes and those that prevent cleavable complexes from forming and later in the same publication suggests three groups based on dose response curves:
Gieseler et al. [1997] suggest a similar set of three modes of drug action:
Table 4. Three categories of topoisomerase inhibitors
| group | Pommier | Gieseler |
| 1 | linear increase non DNA binding |
direct binding to enzyme |
| 2 | bell curve DNA intercalators |
intercalators, cleavable complex |
| 3 | linear decrease bulky intercalators |
inhibit topoisomerase-DNA binding |
One sign of the continuing debate in classification schemes is the recent SAR study of etoposides which models the agents intercalated into DNA [Cho 1996] in contradiction to these schemes and consensus of opinion. The mutation structure analysis presented in this work identifies an allosteric site on topoisomerase unique to etoposide.
Differentiating inhibitors of 2
a and 2b2
a is present in all eukaryotes while 2b is only in vertebrates [Tan 1992]. 2b appears to be more associated with cell differentiation than proliferation [Zwelling 1988, Gieseler 1996]. Decatenation assay with topoisomerase 2 shows two optimal buffer pH peaks, 7.9 and 8.9. The activity at pH 8.9 (2a ) is inhibited by daunorubicin, doxorubucin and etoposide while the other activity at pH 7.9 is not. 2a is clearly inhibited by topoisomerase drugs, but death of cells is better correlated with 2b ; cells with high 2b survive treatment [Gieseler 1997b].Considering the evidence that one potential role of 2
b is as a drug resistance response [Khelifa 1994] it would be worth pursuing these differences in drug development. In line with this thinking, it was recently reported that etoposide, doxorubicin and mitoxntrone have greater effects on 2a than 2b while amsacrine has comparable effect [Meczes 1997]. The temperature sensitive topoisomerase cell model created by the Meczes group ought to help differentiate the roles of 2a and 2b .agent sensitivity |
2a |
2b |
| novobiocin | 200uM |
200uM |
| teniposide | 3x |
1x |
| merbarone. | 8x |
1x |
Drug |
yeast t2 |
human 2a |
human 2b |
human a/B sensitivity ratio |
yeast/human B sensitivity ratio |
| etop | 79 |
23 |
77 |
3.3 |
1 |
| dox | 10 |
2 |
9 |
4.5 |
1 |
| mitoxantrone | >50 |
7 |
>50 |
>7.1 |
1 |
| mAMSA | 38 |
8 |
10 |
1.2 |
1/4 |
| merbarone | 70 |
>500 |
235 |
<0.5 |
3 |
| suramin | >1000 |
>1000 |
>1000 |
1.0 |
1 |
For 3 of the 4 intercalators 2a is several fold more sensitive than 2B. For these same three, the ratio of yeast t2 to human t2B is 1, indicating yeast t2 is more analogous to human 2b than 2a. [Meczes 1997].
The homology model presented in this work of topoisomerase 2
a adds some insight into the active site.Amonafide & Azonafide background
Amonafide was first reported in 1980 as a result of hybridizing the structures of four known cytotoxins. They modeled after the naphthalene from aristolochic acid, the tertiary amine from tilorone and the glutarimide from cycloheximide and CG-603.
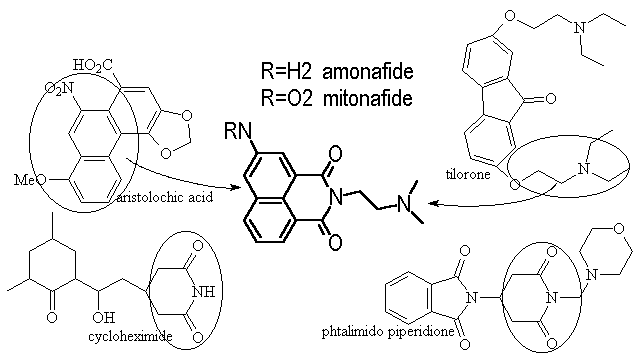 Figure 6 hybrid
origin of amonafide
Figure 6 hybrid
origin of amonafide
Amonafide was immediately found by them to have strong cytostatic activity against both HeLa and KB cells at the sub mM level, just moderate toxicity towards both mice and rats (LD50 >6 mg/kg IP), and the interactions with target site are non-covalent. [Brana 1980] A few years later, unwinding measurements showed it to be a DNA intercalator with the side chain required for activity, and the mechanism is by trapping topoisomerase onto DNA [Hsiang Y 1989]. Clinical trials, however, demonstrated dose limiting myelo-suppression. [Legha 1984; Leiby J; Saez 1989] A review of 15 phase II studies showed hardly any positive results (except for a 14% partial response in breast cancer). [Weiss 1994] Toxicity of amonafide is related to N-acetylation of the aromatic amine [Ratain 1995]. In pursuit of a similar topoisomerase inhibiting compound which will maintain the ability to intercalate but not have the problematic aromatic amine, the anthracene variant of amonafide was developed and named azonafide [Sami 1993].
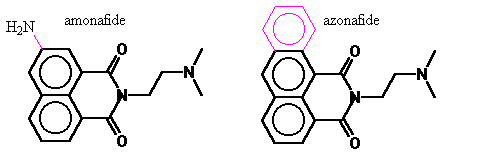 Figure 7. Azonafide
Figure 7. Azonafide
Simply, azonafide can be viewed as a modification of amonafide’s 7-amine replaced with a third planar ring. In addition to avoiding N-acetylation it also is anticipated to have the advantage of larger aromatic electron field to interact to a greater extent with the DNA base pairs. Under the guidance of my advisor, I carried out computer modeling to quantify the difference in DNA binding of these two and model of the mechanism of inhibition.
The goal of the molecular modeling done here is to advance the understanding of topoisomerase and its inhibition to aid in the design and discovery of new and better trapping agents. Due to the a two pronged approach: top down of receptor structure and bottom up of topoisomerase trapping agents, this publication is so divided.
Copyright © 1998 Soaring Bear; Please send your comments bear@dakotacom.net