 Figure 21. DNA
docking gap
Figure 21. DNA
docking gapINHIBITOR METHODS
building molecules:
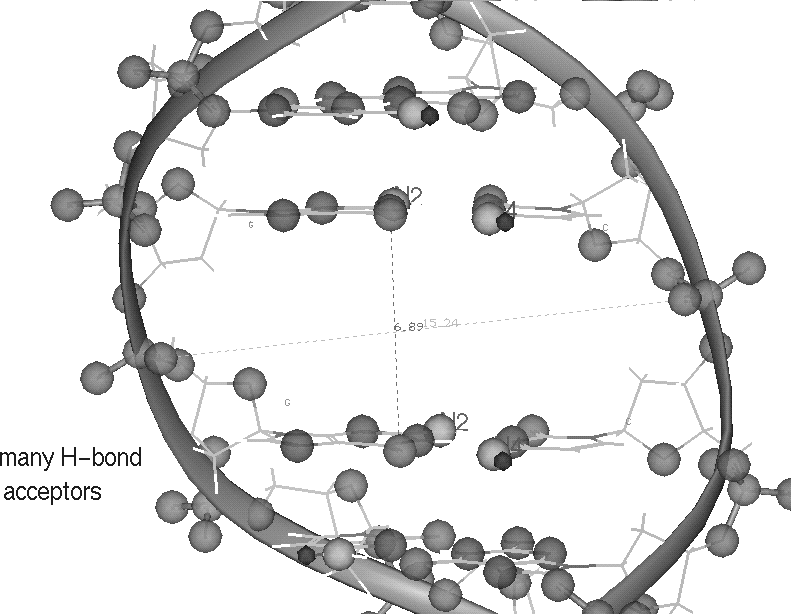
The coordinates data file for the decanucleotide model GGCCGGCCGG.CCGGCCGGCC was kindly supplied by Michael Corey (Burroughs- Wellcome Co.). It contains a gap suitable for manually docking an intercalating drug. This inter-nucleotide gap is located at the midpoint, between G5 and G6 and has an inter-nucleotide distance of approximately double the normal 3.4 A distance. Compounds were constructed with the Build module of Chemlab II program. Coordinates from the minimized structure were used to calculate the partial atomic charges with GAUSSIAN 80 on a VAX. These charges were assigned to the molecule which was then refined in AMBER 3.0 using the following: a distance dependent dielectric constant, a cutoff distance of 99 A for non-bonded pairs, and updating of the pair list every 100 cycles until the root mean square gradient was less than 0.1 kcal/mol A. MIDAS was used to dock the minimized compound into the center of the DNA gap. This was done for both major and minor grooves and with the drug flipped up and down, in both 3' and 5' directions (with respect to the first strand).Calculation & enthalpy comparison: When the compounds are placed into intercalated position in the DNA the side chain at the center, which is a protonated amino hydrogen bond donor, is approximately equidistant from several acceptors: four phosphates of the DNA backbone and O6 of guanine.
This type of docking requires a search of conformational space, representing the range of possible starting positions. To reduce the number of starting configurations a strategy of high enough temperature and dielectric to overcome local minima was used. The idea was to take it to the edge of volatility without crossing over to instability and decomposition of structure. Determining just the right combination of settings which allowed enough looseness of atom movement to search conformational space but enough tightness to keep the hydrogen bonds from falling apart required repeated attempts but was instructive in how unstable DNA is. Dielectric (the distance- dependent reduction of charge-charge interactions) and temperature (Newtonian velocity of the atoms) were the primary adjustments, using the resulting integrity of the helix as a measure of success. Others have suggested improved simulation of water when doing in vacuo calculations by using a dielectric of 4 [Daggett 1991], but I found that reduced the inter-base interactions too much causing the DNA to dissociate and collapse. Consequently, a dielectric of 3 was used for these studies.
The endpoint used was the making of a hydrogen bond between the compound’s tertiary amine side chain and one of the nucleotide acceptors. After a first calculation the drug was re-docked as far as possible in the opposite direction to test if the same result would be reached (i.e. if the first ended to the right, it was re-docked to the left.)
For the solvation dynamics, in order to determine what is sufficient equilibration of water without wasting valuable computer time, a simple water motion evaluation script was made. The script simply gets the X-Y-Z coordinates of each oxygen atom of two files (before and after dynamics) and evaluates the distance moved. It provides the average distance and a rough distribution of the movement by the water oxygens. This was used to assist in developing the combination of temperature and time.
Early results showed dominance of hydrogen bonding of the compound’s amine to DNA backbone phosphates. This is in part because in vacuo calculation lacks salt counter-ions to offset the phosphate charge. Several modifications of the phosphate charges were tried. Greatest success was with Hingerty's method of reducing the electron charge of the each backbone oxygen by 1/4, from -0.847 to -0.597 to bring the phosphate group to half the AMBER standard value and this is what was used throughout.
Calculations were done with AMBER version 3 on Silicon Graphics model 4D/80. Coordinates were captured and structures minimized in AMBER to a RMS of 0.1 kcal/mol. The regime finally decided on was:
CoMFA comparison with force fields: The following operations were performed on a SGI workstation: a) structures were built with Sybyl 6.3 (Tripos Inc), coordinates for the HIVP set were kindly provided by Kate Holloway. b) partial charges were assigned with AMPAC AM1. c) structures were aligned based on the common portion of the molecular set (the Thrombin set was aligned on the 3 atoms which hydrogen bond to the protein). d) CoMFA was conducted with a variety of Tripos and Grid version 15 [Goodford 1985] probes and lattice dimensions and step sizes. e) PLS (Partial Least Squares) was done to determine the important components and predictive error of the model.
Apex QSAR: azonafide analogs were built in Insight (MSI Inc), conformers were generated, cluster was used to reduce the number to under 20, and the results were entered into an Apex database which creates super-impositions and calculates regression of activity associations with chemical features.
Ludi idea generation: this is a software program (MSI Inc) which is basically an idea generator for modifications to a core ligand docked in a receptor. It analyzes a receptor for potential hydrogen bond and lipophilic sites and uses its library of about 1000 fragments to add substituents onto a user supplied core ligand.
Copyright © 1998 Soaring Bear;
Please send your comments bear@dakotacom.net