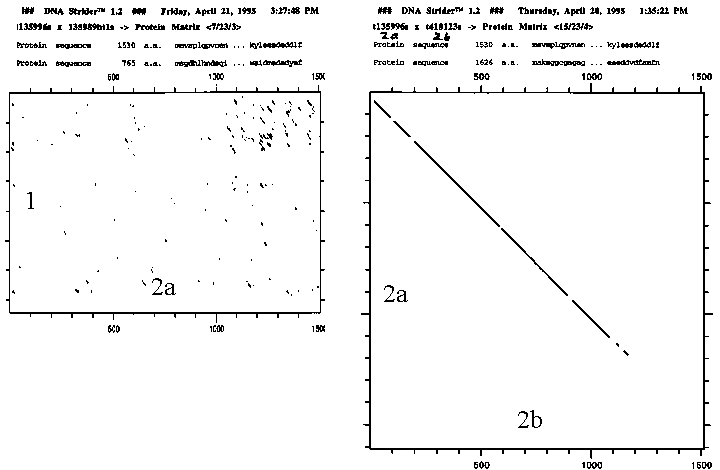
 Figure 9 sequence
matrix human types 1 vs. 2a and 2a
vs. 2b
Figure 9 sequence
matrix human types 1 vs. 2a and 2a
vs. 2b STRUCTURE RESULTS AND DISCUSSION
Sequence analysis of human topoisomerase
Figure 9 sequence
matrix human types 1 vs. 2a and 2a
vs. 2b
Types 1 and 2 are clearly distinct in sequence. Yet it is interesting that the crystal structures for these two (PDB codes: 1ecl & 1bgw) indicate both have an overall donut shape large enough for DNA to fit inside.
Types 2
a and 2b have substantial similarity in sequence. Yet, they differ in some significant ways: 96 residues in length, in location, in level, and in pH optimum for activity. A closer look at the differences is necessary before considering differences in inhibition.Simply from the number of amino acid residues it is clear that topoisomerase is larger than current crystallization techniques allow for obtaining structure. In addition, the function of rotating DNA indicates a very dynamic situation so they were unlikely to be stable globular proteins. It was a pleasant surprise that some partial structures have recently been submitted to Brookhaven PDB. Sequence analysis is still valuable for helping fill in gaps of understanding.
My Blast sequence comparison of 2a and 2B using default blossom 62 (through through expasy: http://expasy.hcuge.ch) shows 81% identity and 91% similarity. Comparison on a percentage basis with each other and the Swissprot average protein shows some interesting things about topoisomerase.
Table 6 2a-2B comparison of sequence
| amino acids | swissprot % 9/97 |
2 a # |
2 a % |
2 b # |
2 b % |
a b diff |
| total | (21 million) |
1530 |
100.0 |
1626 |
100.0 |
|
| Ala (A) | 7.6 |
72 |
4.7 |
95 |
5.8 |
+23 |
| Arg (R) | 5.2 |
68 |
4.4 |
63 |
3.9 |
|
| Asn (N) | 4.5 |
77 |
5.0 |
79 |
4.9 |
|
| Asp (D) | 5.3 |
102 |
6.7 |
118 |
7.3 |
+16 |
| Cys (C) | 1.7 |
13 |
0.8 |
17 |
1.0 |
+4 |
| Gln (Q) | 4.0 |
57 |
3.7 |
55 |
3.4 |
|
| Glu (E) | 6.3 |
122 |
8.0 |
126 |
7.7 |
|
| Gly (G) | 6.8 |
84 |
5.5 |
105 |
6.5 |
+21 |
| His (H) | 2.2 |
24 |
1.6 |
26 |
1.6 |
|
| Ile (I) | 5.8 |
84 |
5.5 |
81 |
5.0 |
|
| Leu (L ) | 9.4 |
124 |
8.1 |
123 |
7.6 |
|
| Lys (K) | 5.9 |
180 |
11.8 |
187 |
11.5 |
|
| Met (M) | 2.4 |
40 |
2.6 |
34 |
2.1 |
|
| Phe (F) | 4.1 |
57 |
3.7 |
72 |
4.4 |
+15 |
| Pro (P) | 4.9 |
74 |
4.8 |
73 |
4.5 |
|
| Ser (S) | 7.2 |
104 |
6.8 |
115 |
7.1 |
|
| Thr (T) | 5.7 |
91 |
5.9 |
89 |
5.5 |
|
| Trp (W) | 1.2 |
18 |
1.2 |
19 |
1.2 |
|
| Tyr (Y) | 3.2 |
43 |
2.8 |
46 |
2.8 |
|
| Val (V) | 6.5 |
96 |
6.3 |
103 |
6.3 |
|
| MW | 174335.2 |
183297.2 |
||||
| pI theoretical | 8.92 |
8.14 |
||||
| neg Asp+Glu | 11.6 |
224 |
14.7 |
244 |
15.0 |
+20 |
| pos Arg+Lys | 11.1 |
248 |
16.2 |
250 |
15.4 |
+2 |
| net charges | -.5 |
+24 |
+1.5 |
+6 |
+.4 |
-18 |
| instability index* | 40.36 |
36.27 |
||||
| Aliphatic index** | 75.92 |
73.14 |
||||
| Ala+Val+Ile+Leu | 29.3 |
24.6 |
24.7 |
|||
| hydropathic*** | -6.920 |
-6.549 |
*based on dipeptides; over 40 is predicted as unstable; [Guruprasad 1990].
**Ala+Val+Ile+Leu mole percent [Ikai 1980]
***Grand average of hydropathicity (GRAVY) [Kyte Doolittle 1982]
Compared with the SwissProt database containing nearly 60,000 protein sequences, topoisomerase has less alanine, half the cysteine; more charged residues; and is overall more positive. The positive charge is in agreement with the function of binding to negatively charged DNA. The instability index corresponds with the spread out arrangement seen in the crystal structure (discussed below) and with the function of swinging cut ends through a change in super-coiling and is in agreement with the half life of only 5-10 minutes.
Some biochemical and pharmacological properties of 2a and 2B have been determined which point up some differences between them [Drake 1989a]
a and 2b comparison of properties| property | 2a | 2b | units |
| growth phase | hi in log growth days 2-3 |
gradually inceasing days 5-6 |
- |
| relaxation kinetics | distributive (relax-dissociate) |
highly processive (relax-relax) | - |
| oligo sensitivity | AT & GC | GC | - |
| suggesting cuts at: | AT | GC | - |
| nuclear localization | nucleoplasmZini | nucleolusZini | - |
| Tm half life @25C | 10 |
5 |
min |
| ATP conc | 1 |
1 |
mM |
| V inhibition | 2 |
2 |
uM |
| KCl | 120 |
140 |
mM |
| novoiocin sensitivity | 200 |
200 |
uM |
| teniposide sen. | 3x |
1x |
|
| merbarone sen. | 8x |
1x |
The more processive character of 2B is suggestive of a stronger DNA binding.
2
a and 2b are similar enough to form a substantial fraction of alpha/beta heterodimers [Biersack 1996].2
a is present in all eukaryotes while 2b is only in vertebrates [Tan 1992]. 2b appears to be more associated with cell differentiation than proliferation [Zwelling 1988, Gieseler 1996]. Decatenation assay with topoisomerase 2 shows two optimal buffer pH peaks, 7.9 and 8.9. The activity at pH 8.9 (2a ) is inhibited by daunorubicin, doxorubucin and etoposide while the other activity at pH 7.9 is not. 2a is clearly inhibited by topoisomerase drugs, but death of cells is better correlated with 2B; cells with high 2B survive treatment [Gieseler 1997b].Considering the evidence that one potential role of 2B is as a drug resistance response [Khelifa 1994] it would be worth pursuing these differences in drug development. In line with this thinking, it was recently reported that etoposide, doxorubicin and mitoxntrone have greater a effect on 2a than 2B while amsacrine has a comparable effect [Meczes 1997]. The temperature sensitive topoisomerase cell model created by the Meczes group ought to help differentiate the roles of 2a & 2b.
Although known about for nearly three decades [Wang 1971] this structure has been resistant to crystallographic determination because of its huge size. Even by expressing a yeast form lacking about 400 residues at each end to get a crystal [Berger 1996], it is still one of the biggest structures determined to date. With this partial structure of about 2/3 of yeast topoisomerase 2 it became obvious that another factor making crystallization difficult are the long hydrophilic arms. Given the 45% identity to human sequence [Expasy], considerable understanding is gained from this structure. Although the authors provided a substantial description of the structure, there were some points which I am able to add after my own structure analysis.
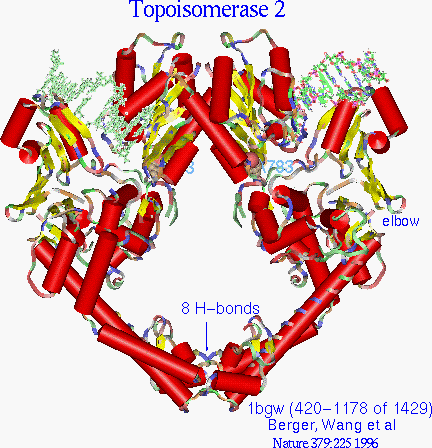 Figure 10.
Yeast topoisomerase crystal has a scissors jack appearance
Figure 10.
Yeast topoisomerase crystal has a scissors jack appearance
The first impression of the yeast topoisomerase structure is of an overall donut shape. Each monomer is roughly a semicircle, and dimerizes end to end to orm a sort of donut with a hole in the center. In contrast to the topoisomerase 1 which has a layer of positively charged residues on the surface of the inside of its donut shape, topoisomerase 2 lacks that distribution and so appears to be less likely to embrace DNA that way. Also a closer look discloses that the donut is not very round. Graphical manipulation of the structure reveals two long arms of extended alpha helices which join like pincers (held by 8 hydrogen bonds) and distinct elbow joints which bring to mind a scissors jack.. Compared with other protein structures, which are usually compact and globular, topoisomerase 2 is a relatively loose, unrestrained enzyme, having lobes and long solvated arms. The hydroxyl of the active site Tyr (783 for S cerevisiae) is coordinated, and presumably held in place, between several charged residues. This will be discussed in greater detail later.
Junctions of the monomers
As can be seen in the figure, there are two junctions of the monomers. Careful examination reveals that the upper junction is dominated by hydrophobic forces and the lower one by hydrogen bonds. Engelman-Steitz hydrophobic analysis [Engelman 1986] (window of 5), reveals a concentration of hydrophobic residues in the bulky upper region. This region, containing the active site tyrosine, is also held by two inter-monomer cysteine sulfur bridges. There are only 2 hydrogen bonds in that whole region. The lower junction is like the meeting of fingers of two long helix arms. The Insight hydrogen bond feature (H-heavy atom distance of 2.5) reveals 8 hydrogen bonds appear in that small area of around 300 A2 (see figure). There are 32 residues in that junction which are unresolved (which hints at motion) so there could be more.
Recognizing that the upper junction of the two monomers is held together primarily by hydrophobic interactions and the lower part by hydrogen bonds begs the question of which might be more strongly held and what conditions might modulate either coupling. There has been much discussion in the literature speculating whether the enzyme operates by a 1 or 2 gate mechanism, so it is interesting to see that the two possible gates are held by different forces. None of the drug selected mutants, discussed below, are in either of these gate areas, suggesting the importance to cell survivability of these contact areas. Further research about this could take advantage of the different forces. The hydrogen bonding would be expected to change with pH and ionic conditions. It has already been established that winding activity of topoisomerase is very sensitive to both of these [Osheroff 1983] but not with the perspective of affecting one gate and not the other. The hydrophobic interactions of the upper part could be modulated by mild detergents. In both cases, the changes would have to be kept in a modest range to reduce changes in folding of the monomers.
DNA docking
Considering the extensive interaction this enzyme has with DNA it is rather obvious to attempt dock an oligomer with this crystal structure. The mutant analysis (next section) and some electron micrography [Schultz 1996]] indicate the upper portion to be the DNA binding region. During test docking I observed that a bulky helix formed by residues 532-548 fits into the major groove better than the minor groove. It is about 15-20 A from the Tyrosine so about a half turn of DNA from the cut. This might be how DNA is aligned properly before cutting by the active site tyrosine so I call it the major groove aligning helix. I was unable to confirm from this single structure the report that topoisomerase preferentially binds to curved DNA [Bechert 1994].
The lobes in the upper portion are positioned well for nesting DNA, forming somewhat of a bore-hole barely large enough in diameter for DNA. The size of the hole tapers down to the active site Tyrosine 783 at the bottom of the hole. The taper would only allow a single oligo-nucleotide strand reach the bottom. It is difficult to say from this whether that attached strand is the overhang or the shorter strand. The distance in the crystal of the catalytic tyrosine hydroxyls is 28 A, far more than a 4 base pair overlap (considering 3.5 A per step). Manipulating the structure to squeeze together the scissors jack elbows can bring the distance down to 14 A, just right for 4 base pair overlap. Since there is only one topoisomerase 2 structure available it is difficult to guess the complexities of movement. But there is something to say about the tyrosine attachment and strand overhang.
The cycle of topoisomerase attachment, cutting, strand passing, re-ligation, and detachment has been elegantly described [Osheroff 1991]. The temporary cut by topoisomerase 2 is accomplished by the hydroxyl of a tyrosine attacking each side of the phosphate backbone, performing a trans-esterification. This results in tyrosine attached covalently to the phosphate and a nucleotide hydroxyl on the cut side. Due to the transitory nature of the cut and the indefiniteness of the twisting endpoint, it is difficult to directly determine whether attachment and overhang are 3' or 5'.. An indirect polymerase extension method indicates 5’ overhang and 5’ attachment (the 5th line of table 7 below), leaving a 3' OH during the transient cut [Liu 1983, Morrison 1979]. The authors did admit to the suggestive quality of this data but the model has been repeated so many times in review publications that this has become dogma which sometimes doesn’t even have a reference cited. The oft repeated crude model appears like this:
tyr
O
|||||||||
||||||
|||||||||| |||||
O
tyr
Figure 11. Four base pair overlap of DNA cut - widely held model
In my attempts to dock DNA into the topoisomerase it didn’t fit right to satisfy this widely held model. By considering how the cut DNA with overhangs would fit into topoisomerase, I found a basis for disagreement with the generally held model.
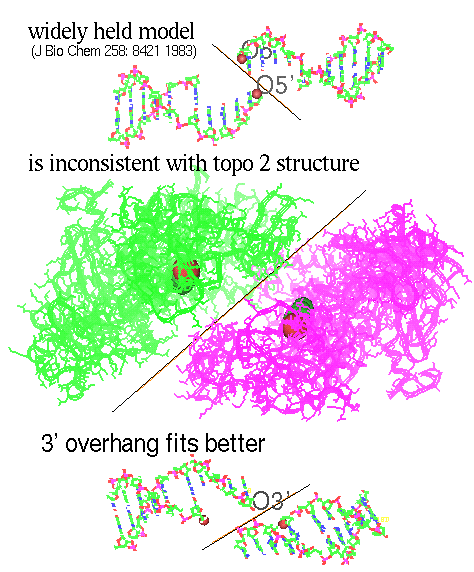 Figure 12. 5’ or 3’ overhang? (top view)
Figure 12. 5’ or 3’ overhang? (top view)
In the figure above, the upper view of oligomer renders the generally held view of a 5’ overhang and 5’ attachment (line 5 of table 7), leaving a 3' OH during the transient cut [Liu LF 1989, Wang 1996]. Notice in the figure how the diagonal of the cut junction fails to match the slant of the meeting of the two units of topoisomerase.
The lower oligomer shows one possibility (line 2 of table 7) with a cut angle which would match the topoisomerase structure better. There are some other possibilities. Theoretically, a pair of cuts in DNA could end up with 8 different combinations of overlap and tyrosine attachment as shown in the table below.
Table 8 Eight types of DNA attachment & overhang
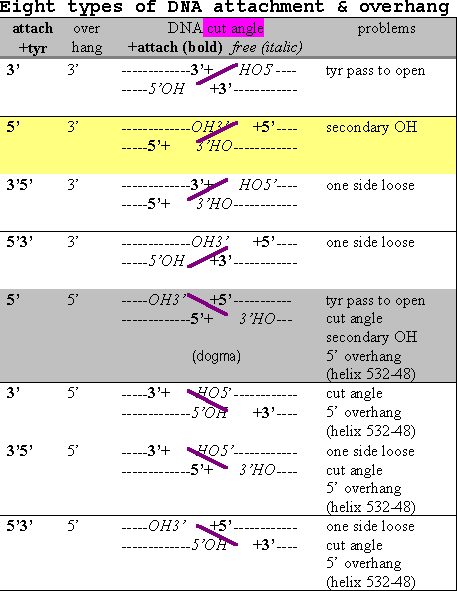
The actual type necessarily depends on how the attacking tyrosines and assisting residues are spatially placed by the enzyme (discussed further in the active site section later). The 4 base pair separation is equivalent to nearly a half turn of the usual B-DNA so the phosphates somewhat face across the groove from each other. The 3’ overhang types would face across the minor groove while the 5’ ones face across the major groove. If we knew which groove, the modeling would be simplified by half and we’d know which to target with inhibitors. A structural steric factor is a bulky helix formed by residues 532-548 at the base of the DNA docking region which fits into the major groove better than the minor groove. The end of the helix, residue 548, is about 15 A from Tyrosine 783, about 5 base pair steps or about a half turn of B-DNA. This would favor the two tyrosines facing across the minor groove and the 3’ overhang. So the enzyme feature might be called a groove determining helix.
I have identified 5 types of conceptual problems which affect the likelihood of the eight possibilities. It is not uncommon for enzymes to overcome some mild energetic barriers but some of the problems noted create substantial obstacles. Interestingly, there are some conceptual problems with all eight types in the table. And most interesting is that the widely held model has the most conceptual problems!
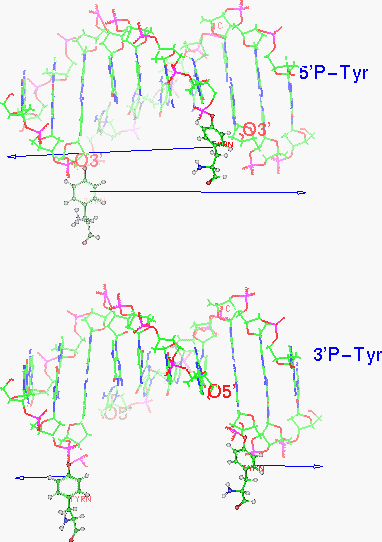 Figure 13.
Tyrosine passage and overhang, lines 5 & 6
Figure 13.
Tyrosine passage and overhang, lines 5 & 6
5’5’ (line 5), the widely held model, is somewhat untenable in requiring the tyrosines to pass each other during separation. 3’5’ (line 6) would be in a better position to separate more easily as seen in the above figure. Another advantage 6 has over 5 is the primary 5’ hydroxyl left free. However, both these types have the common problem of the cut angle not fitting the crystal structure (as described above).
Lines 3,4,7 and 8 have in common the unlikely arrangement of two covalent attachments to one side and none to the other leaving one piece of double strand DNA loose and an asymmetric cut by an otherwise symmetrical homo-dimer.
Lines 1 and 6 would be thermodynamically favored over lines 2 and 5 due to leaving a free 5’ (primary) OH.
In conclusion, modeling the structure provides substantial doubt about the widely held dogma. The 5’3’ (line 2) cut has the smallest structural obstacle, with 5’ attachment and 3’ overhang. Great advances in the tools of molecular biology have been made in recent years and it is hoped that new methods be used to revisit this matter to clear it up.
Drug selected mutant bioinformatic analysis
A variety of mutant topoisomerase have been reported from drug resistant cell cultures and from site directed mutagenesis studies. Without a structure, the mutants can only be laid out along a simple linear sequence as was done in recent papers [Rubin 1996; Beck 1996]. Without relating the location of these mutations relative to active site and other structural components only minimal insight is gained. A quantum jump in understanding the enzyme can be gained by identifying where on a three dimensional structure these mutations are. Mutations affecting drug action are instructive in the dynamics of the structure.
It has been shown that human leukemia cells selected for m-AMSA resistance, which have Arg486>Lys mutation, are not cross resistant to other topoisomerase inhibitors (etoposide, doxorubicin) [Lee 1992]. This is a conserved ATP binding motif [Hanks 1988; Wyckoff 1989]. The homologous mutation in yeast topoisomerase 2, Arg476>Lys has been reported without AMSA resistance while change in the adjacent residue 475 does confer resistance [Wasserman 1994]. E. coli gyrase, which has Lys at the homologous position is resistant to m-AMSA and the mutation, Lys447>Glu is responsible for nalidixic resistance [Yamagishi 1986].
The literature of the mutants is summarized in the table below and analyzed with respect to the three dimensional structure in the discussion afterward.
Table 9 drug induced mutations
| Mutation | amsa | etop | ellip | dox | mitox | Fquin | cmpt | drugs | ref |
| Sc Gly144>Ile,Pro lose ATPase |
Lindsley | ||||||||
| cho C426>Y | R | Rizvi | |||||||
| Consv 429-56 ATP[Bugg] | |||||||||
| GxxGxGKTxxxxxx(I/V) | |||||||||
| hu Ala429>del | r | R | Campain | ||||||
| Sc Lys439>Gln | r | r | Nitiss | ||||||
| Sc Lys439>Glu | r | r | Nitiss | ||||||
| hu Arg449>Gln & Pro802>Ser =Sc439 |
r R | Danks | |||||||
| hu Arg449>Gln | R | Bugg | |||||||
| hu Arg450>Gln or 803 | r | r | Hsiung Nitiss | ||||||
| Conserved 474-81(Sc) PLRGKMLN, ATP motif [Wasserman, Wyckoff] | |||||||||
| hu Arg486>Lys | R | Lee | |||||||
| hu Arg486>Lys | r | Zwelling,Hinds | |||||||
| hu Arg486?> & ?494>? | R | Kubo | |||||||
| gy Lys447 =hu486 | r | HindsYamagishi | |||||||
| gy Lys447>Glu =hu486 | r | Yoshida | |||||||
| Sc Arg476>Lys | = | Wasserman | |||||||
| Sc LR475-6> AG | r | Wasserman | |||||||
| Sc PLR474-6>AVG | r | Wasserman | |||||||
| Sc Lys478>Ala | r | Wasserman | |||||||
| cho Arg493>Gln | R | Chan | |||||||
| Sc His507&521>Tyr A4 | r | r | Nitiss | ||||||
| Sc Ala642>Thr/Gly Consv | r R | Wasserman | |||||||
| Sc Ala642>Ser | R | Patel 97 | |||||||
| Conserved 735-41 [Liu] | |||||||||
| gy Ser83>Ala =Sc741 | r | Reece | |||||||
| gy Ser83>Leu =Sc741 | r | Reece | |||||||
| gy Ser83>Trp =Sc741 | r | Reece | |||||||
| Sc Gly738>Asp 101* | = | r R | r | Nitiss,Liu | |||||
| Sc Ser741>Ala =gy83 | = | = | = | = | Nitiss | ||||
| Sc Ser741>Leu =gy83 | = | = | = | = | Nitiss | ||||
| Sc Ser741>Trp =gy83 | = | + | r | Nitiss Hsiung | |||||
| Sc Gly748>Glu | R | Patel 97 | |||||||
| hu Ala744>Thr | R | Takano | |||||||
| hu Arg772>Ile | R | Danks93b | |||||||
| hu Lys797>Asn | R | Patel | |||||||
| Consvd 803-4 RY | |||||||||
| hu Pro802>Ser or 450 | r | r | Hsiung Nitiss | ||||||
| hu Pro802>Ser (& Arg449>Gln) |
r R | Bugg, Danks | |||||||
| Conserved 818-30 [Liu] 829-39 NGAEGIGTGWS nucleotide binding |
|||||||||
| Sc Pro824>Ser (& Gly1186>Glu) 103* |
r | r R | = | Nitiss Liu | |||||
| Sc Pro824>Ser | r | r | = | Liu | |||||
| Sc Pro821>Gln @25 4 | r | Holm Liu | |||||||
| cho Gly851>Asp =Sc830 | R | Hashimoto | |||||||
| hu Ser861>Phe | R | Kohno | |||||||
| Sc Arg884>Pro RYRM>PYII 5 | r | r | r | r | r** | Nitiss Jannitipour | |||
| Sc Gly1186>Glu | = | = | Liu | ||||||
| Sc Arg1195>Lys 102* | r | r | r | Nitiss Liu | |||||
| cho | r | Rizvi,Ng | |||||||
| Conserved 1012 | |||||||||
| Sc His1012>Tyr arm | = | r | + | R | Froelich | ||||
| Sc t2-1 temp sen @25 | r | Nitiss | |||||||
| Sc t2-1 temp sen @30 | + | Nitiss | |||||||
| Mutation | amsa | etop | ellip | dox | mitox | Fquin | cmpt | drugs | ref |
notes: r resistant, R drug selection, rR drug sel confirmed by site dir mut;
= unchanged
+ Sensitive
* reconstructed 101 has normal drug response, lack explaination;
knot activity: 102 normal, 101 10%, 103 25% of normal wt [Liu]
** increased cleavage rather than inhibiting re-ligation (30,31)
Interestingly, all of these drug induced mutations are relatively distant, over 10 A, from the catalytic RYP. In regards to that it is worth recalling this is an extended flexible protein and all distances are subject to change. The differences in how particular sites affect sensitivity to different drugs gives some insights in varying mechanisms of the different subgroups of topoisomerase 2 inhibitors. Structure activity relations will be discussed in greater detail below but there is much confusion in that and some clarification may be gained by combining that information with this mutant map.
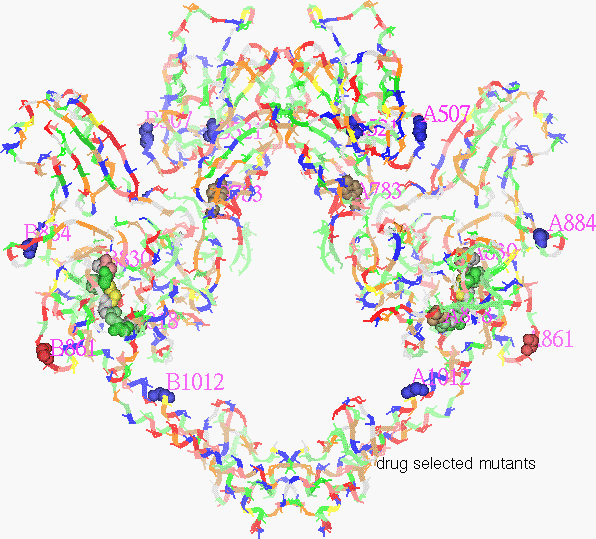 Figure 14.
Residues 1012, 861, 818-30, 884, 521 & 507
Figure 14.
Residues 1012, 861, 818-30, 884, 521 & 507
Most striking is the mutation at residue 1012 (His to Tyr), near the joint of the helical arm extension. This change makes cells resistant to etoposide, sensitive to ellipticine and unchanged to amsacrine. The exact mechanism is not incontrovertible for either of these compounds but it is widely believed that the first is a direct binder and the other two are intercalator poisons. The sensitivity to the intercalators, amsacrine and ellipticine, demonstrates continued functionality of topoisomerase. So this change apparently doesn’t disable topoisomerase but mainly changes etoposide inhibition. Some speculation might be made of the implication of this based on the differences in properties of histidine from tyrosine, most obviously size, and pKa. The larger aromatic tyrosine might provide steric protection from binding by etoposide polar groups. The higher pKa of tyrosine, 10 vs. 8 might affect hydrogen binding or transfer.
The Ser861>Phe is also by the arm joint. My multiple sequence alignment of 115 topoisomerase sequences shows it to be well conserved except for being replaced by alanine in human 2B, Crithidia , and Trypanosoma , or lysine in African swine fever. This might have implications in isomer or species selective inhibition.
818-30, also near the arm joint, is conserved in Saccharomyces, T4 phage, Drosophila, human 2a, and gyrase [Liu]. Affects of changing Pro 821 or 824 suggest some structural importance for prolines here, which form a beta turn with the two intervening residues. Changing proline 824 causes resistance to both amsacrine and etoposide but not to quinolone whereas changing proline 821 does impact the quinolone.
Arg884>Pro creates resistance to all five compounds tested indicating fundamental importance. It is on a lobe which has been hypothesized to cradle the DNA being cut. It is common for positive amino acids of DNA binding proteins to form charge interactions with the negative DNA. So it appears that this arginine is involved with topoisomerase binding to DNA.
His507&521>Tyrosine is one of the closest to the catalytic Tyr783, at 16 A away. Although some distance from the active site, these are among the closest of histidines to the active site and might be involved in a proton transport chain for the proton which moves for the trans-esterification reaction.
 Figure 15.
Residues 429-81, 735-41
Figure 15.
Residues 429-81, 735-41
429-56 [GxxGxGKTxxxxxx (I/V)] is a conserved region and all known mutations in this segment resist drug activity. The closest point of this segment to Tyrosine 783 is over 20 A.
474-81(Sc) PLRGKMLN is an adjoining lobe and also conserved region and identified as an ATP motif [Wasserman 1994, Wyckoff 1989]. The only mutant in this group that didn’t cause drug resistance was the very conservative arginine to lysine change. The Gly144>Ile or Pro losing ATPase activity suggests it might loop over to somewhere near this region. Although change at residue 1186 has no effect on drug resistance, even a conservative arginine to lysine change at the nearby residue 1195 makes the enzyme resistant to all three compounds tested. Although the crystal stops at 1178, that is part of a long helical arm which reaches up toward residue 439.
The section, 735-41 (Tyr-His-His-Gly-Glu-X-Ser) is conserved in Saccharomyces, Drosophila and human 2a (751-62) and similar in gyrase [Liu]. The Gln740 is 15 A from Tyr783. Replacement of Gly738 by the larger aspartic acid makes topoisomerase resistant to etoposide, amsacrine and fluoroquinolones, indicating a fundamental effect. The multiple changes done at Ser741 give a little insight to that site. Changing to polar alanine or non-polar leucine has no effect while a bulky tryptophan causes sensitivity to etoposide, resistance to fluoroquinolones and no change for amsacrine.
A study with a mutation only vaguely identified as being in the 3’ region, is nonetheless instructive about selective resistance to drugs. A MDA-VP human breast cancer cell line selected by exposure to 1 ug/ml etoposide had a drug survival fraction which increased 20-60 fold with etoposide but only 10 fold with amsacrine and 2 fold with doxorubicin [Asano 1996b]. This corresponds with a prior study selecting human leukemia cells with amsacrine which developed a 208 fold resistance to amsacrine and only 6 fold for etoposide [Lee MS 1992]. This latter study also showed only 3 fold resistance to doxorubicin, which is commonly believed to intercalate DNA and poison topoisomerase much like amsacrine, suggesting differences even between intercalators.
Further proof of the impact of topoisomerase mutation is provided by a few studies using gene rescue. Etoposide resistant human brain tumor cells were re-sensitized by transfer of Drosophila topoisomerase II gene [Asano 1996a] or human topo2a gene [Asano 1996c]. Etoposide selected human breast cancer cells were re-sensitized by human topoisomerase 2a [Asano 1996b]. Etoposide resistant Chinese hamster ovary cells became 20 fold more sensitive to the drug when Drosophila topoisomerase 2 was transfected and even though the Drosophila t2 mRNA was only 0.1% of endogenous mRNA [Eder 1993].
Although this bioinformatic sequence-structure analysis lacks definitive finding on its own, it does contribute to seeing that there are multiple ligand inhibition sites on topoisomerase and multiple modes of poisoning. In overview, it is interesting that the area within 15 A of the somewhat embedded catalytic tyrosine is relatively free of mutations. Most of the drug resistant mutations are over 20 A away from there, and appear to have more to do with the binding of DNA or the kinetic flexing of enzyme extensions. The two adjoining lobes formed by residues 429-456 and 474-481 account for many of these mutants. It is unfortunate that etoposide and AMSA so dominate these studies. The hard work of selecting these cell mutants has already been done and it would be relatively trivial to test on them the other agents to fill out the chart and provide more insight of drug selective effects.
Homology of human topoisomerase structure from yeast
It is generally considered that sequence identity of over 25% makes a candidate for creating a homology model to reveal evolutionary changes which might offer insight to species selective inhibitors. The recently elucidated Saccharomyces cerevisiae yeast topoisomerase 2 [1bgw, Berger 1996] , which has 45% identity to human 2a and 46% to human 2B [http://expasy.hcuge.ch] made a good candidate.
Aside from the Pro802 mutation , just two residues from the active site tyrosine, the closest amino acid change to the active site tyrosine is a histidine (H521), 12 Angstroms away [Nitiss 1994].
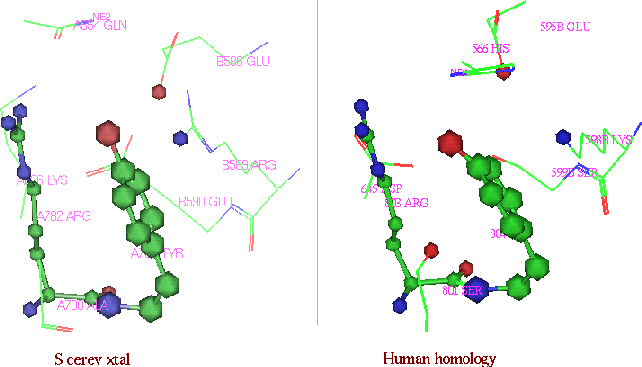 Figure 16.
Active site-yeast vs. Human
Figure 16.
Active site-yeast vs. Human
The active site figure above shows the substantial similarity of the yeast crystal structure with the human homology model with the most significant change being yeast glutamine 554 evolved to His 566 in human. That could make a difference in pKa which ought to be significant considering the enzyme tyrosine attack involves a proton transfer. This change suggests evolution to a better proton donor for the trans-esterification reaction with the DNA phosphate. Histidine often appears near the active sites of enzymes which assist proton transfer (reductases, dehydrogenases, endonucleases) because it is the amino acid with the closest pKa to physiological pH. Histidine can provide a pathway for protons to be transferred to the active site. With other enzymes, it has been found that mutating an active site histide to glutamine can reduce activity; as evident with dehydrogenase [Ehrig], xylose isomerase [Lee], acetyltransferase [Lewendon], phospholipase [Annand], and glycohydrolase [Antoine]. Yet, the yeast topo has functioned for a billion years, so it apparently uses the acidic glutamates for proton transfer rather than a histidine imidazole. Proteins which use glutamate for proton transfer include: RNase [Gohda], rhodopsin [Jager, Brown], and photosynthetic reaction center [Paddock]. Glutamine has been identified as part of the proton transfer path in triose-phosphate isomerase [Joseph-McCarthy] and cytochrome [Paddock]. A change in route of proton pathway, from glutamate to histidine, could help explain why topoisomerase inhibitors used for cancer therapy are not necessarily anti-fungal, and vice versa.
The tyrosines involved in the trans-esterification, yeast 783 and human 804 are shown in the figure in bold towards the viewer. For both species, the tyrosine oxygen which attacks the phosphate is held between two charged nitrogens, 3.5-4.7 A to either side. On the left is an adjacent arginine (a-R782), which is conserved in the human model (a-R803). On the right is an arginine of the other (beta) monomer (B-R589), which is conservatively replaced by a lysine (B-K598) in humans. In the crystal the tyrosine topoisomerase is in a pinched area of the enzyme, with the hydroxyl right at one surface (presumably where DNA binds) and only a couple of amino acids from the other side of the enzyme. This closeness to the inside surface of the donut shaped crystal leads to speculation of alternate DNA binding models.
Away from the viewer, deeper and behind the tyrosine is a glutamic acid from the other monomer (B-E586), holding its oxygen just 2.7 A away from the Tyr-O. The closeness suggests a shared hydrogen in between. This glutamic acid is conserved in the human as B-E595. Nearby is a trio of glutamic acids (B-E590-2) which are embedded and form something of a path to the inner surface of the donut shaped crystal. This acid path has been replaced in humans to hydroxyl path of Ser-Ser-Thr. This might possibly be a proton transport path and the evolution to hydroxyls could be expected to slow such transport.
There is coordination between two basic amino acids and two acidic ones which form somewhat of a box of about 5-7 angstroms on each side.
Slightly further away, 5.4 A from the Tyr-O, is the carbonyl of Asp 552 which holds it’s side chain carboxyl out into solution. It is a candidate residue for coordinating the magnesium ion ; however, it has evolved in humans to arginine 565 which would repel a Mg2+ but provide stronger interaction with DNA negative charges. Further up along the surface, are glu-asp 512-513 which are conserved in human as glu-asp-glu-asp 522-5. They are at the tip of a floppy loop which, in the crystal, is at the base of the major groove determining helix (532-48). The doubling to 4 acid residues in human topoisomerase might make for better Mg2+ coordination. Also nearby, and possibly contributing to Mg2+ coordination, is Asp 635, conserved as Glu-Asp 643-4 in the human sequence.
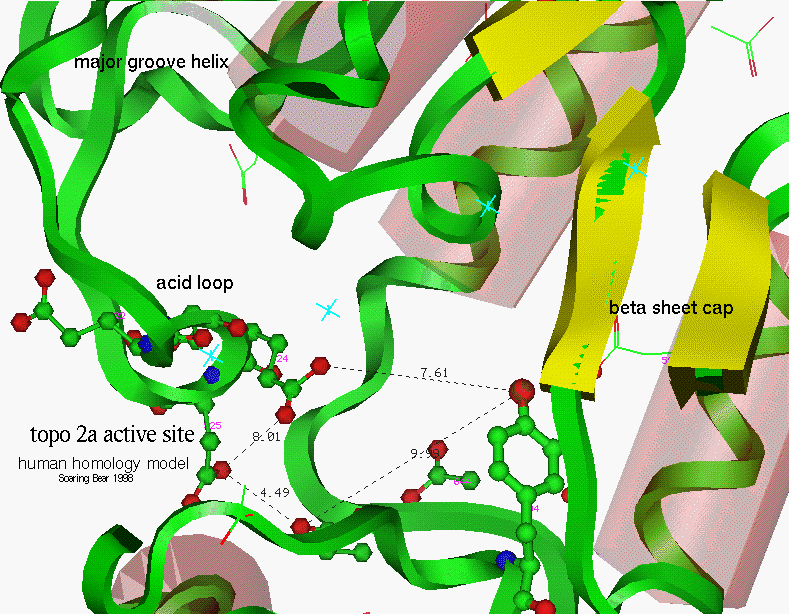 Figure 18
Active site, human 2a
Figure 18
Active site, human 2a
Looking further away from the active site tyrosine, the nearest histidine in the yeast topoisomerase structure is 12 angstroms away. Three others are (in different directions) within 15 angstroms, Interestingly, the nearest histidine, H521, mutates to tyrosine in etoposide resistant cells [Nitiss].
In conclusion, human topoisomerase could be interpreted as an improved form in view of the His 566 being a better proton donor than yeast Gln 544 and the acid repeat at 522-5 providing more control of the Mg2+ positioning than yeast 512-3. The Asp 552 in yeast is well positioned to fit in the major groove of DNA and evolution to Arg 565 might be to improve interaction with negatively charged DNA or to reduce susceptibility to intercalator topoisomerase poisons.
It would be worth following this homology model by creating one of topo 2B to pursue the sequence comparison described earlier.
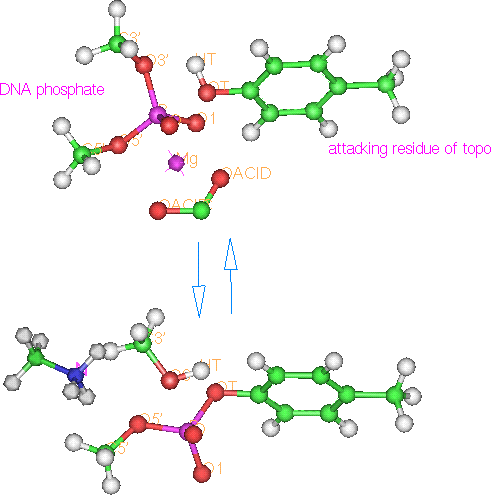 Figure 19.
Trans-esterification reaction
Figure 19.
Trans-esterification reaction
The above reaction is readily reversible and there are some calculations which suggest the change free energy is quite small, on the order of 1 Kcal/mol [Champoux 1990]. Semi-empirical calculations lends support to the modest energy. In fact, implying that the broken phosphate is actually favored over intact phosphate by a little more than one Kcal/mol. This suggests a reason why ATP is required for the final re-ligation step and not for the initial breaking step.
Table 10. Transesterification energy
| Method | Kcal/mol |
| AM1 | -1.5 |
| PM3 | -3.7 |
| MNDO | -3.0 |
Magnesium is required to make the cut in DNA but it is not clear whether or not it is involved with the reconnection [Osheroff 1991]. Magnesium has been observed crystallographically 1.9 A from DNA phosphate oxygens for E. coli RV endonuclease coordinated between oxygens (2.3 A) of Asp 74 and Glu 45 [Kostrewa 1995]. It is possible that Mg2+ cation assists the cutting reaction by distorting the electron density of the phosphate to allow closer approach of the tyrosine for nucleophilic attack. My Spartan calculation with Mg ion provides a model of how the cation can distort the electron field.
Figure 20. Mg ion (lower) pulls phosphate electron density down
As seen in the figure, one would expect the Mg2+ induced distortion to also help determine which of the phosphate bonds is broken and, therefore, whether the tyrosine is attached 5’ or 3’, leaving the other as free OH. Since there is no obvious histidine proton donors nearby it can be conjectured that the proton for the free OH comes from tyrosine 783 as it’s oxygen attacks the phosphorus.
One can also anticipate that said proton will be repulsed from the Mg2+ so if the Mg2+ is held near the 5’ oxygen the proton would be nearest to and protonate the 3’ oxygen, resulting in a 5’ attachment, as with types 5 and 2 in the earlier discussion, the dogma and my preferred ones, respectively. While this doesn’t in itself resolve the attachment issue, it does contribute to narrowing down the other possibilities. It is ordinary for catalytic cations in an enzyme active site to be coordinated and guided by acid residues. Examining the region near the active site Tyr-O reveals only a few acidic residues, even though the sequence analysis shows a higher than average presence of them (15.7% vs. 11.6%) for the whole protein [Expasy].
In the yeast crystal structure Asp 552 is held out alone into solution about 7 A from the tyr-O and so could very well receive the Mg2+ ion. It can be speculated that the Mg2+ will be coordinated at the interface junction of the topoisomerase and DNA between Asp 552 and a phosphate. A test docking with B-DNA into the active site holding a backbone phosphorus a few angstroms from the Tyr-O demonstrated the feasibility of an Asp 552 oxygen being 5 A away from the O5’, about the right distance to have an intervening Mg2+. In this test configuration the Asp 552 is in the major groove, less than 5 A from a Gua N7 1 ½ steps up the DNA from the Tyr-O. The side chain of an intercalator docked two bases away from the tyrosine cut is well positioned to interfere with the Asp 552. However, Asp 552 has evolved to Arg 565 humans so could explain some species difference in enzymatic activity and selectivity which might be exploited in drug design.
The next nearest acidic residues are the pair Glu-Asp 512-13. They are not only conserved in human topoisomerase but also repeated as Glu-Asp-Glu-Asp (522-5). They are well positioned to be along the DNA backbone and might coordinate the required Mg2+ with phosphates. These residues are on a floppy loop which is in contact with the end of the groove positioning helix (532-48 in yeast).
Further elucidation of this would involve site directed mutations on these acid residues. Alanine ought to eliminate activity. It might also be interesting to mutate the potential proton transport path (acidic 590-2 in yeast or 599-601 in human) mentioned earlier to see if that stops proton transfer of trans-esterification.
That inhibitors interfere with reconnection and not with cutting suggests some kind of interference with the ATP hydrolysis. A conserved segment of the topoisomerase 2 sequence (474-81 in yeast), which has been suggested to be an ATP binding motif, has been shown in the mutant analysis (described earlier) to be associated with drug resistance. Too much of the region near this is unsolved to make further statements about it.
The topoisomerase-DNA cleavage complex with inhibitor is sometimes referred to as the ternary complex. Considering the requirement of ATP and Mg, it might just as well be called the quinary complex
This concludes the top down analysis from the perspective of macromolecular topoisomerase and DNA structure effects on inhibitor design. Discussion will now shift to the bottom up approach of looking at the structure-activity relations of inhibitors.
Copyright © 1998 Soaring Bear; Please send your comments bear@dakotacom.net